PHERAstar FSX
Powerful and most sensitive HTS plate reader


Barry Whyte is Application Scientist and Science Writer at BMG LABTECH in the United States. He has PhD and Bachelor of Science (BSc) degrees in biochemistry from the University of Bristol in the United Kingdom and more than 20 years of experience in the life sciences and science communications. Over the years, Barry has worked on three continents and traveled widely. He enjoys building on his international work experience and learning new ways to help scientists advance their research.
Interest is growing in the use of targeted-protein degradation to induce the breakdown of disease-causing proteins after treatment with small molecule drugs.1 Targeted protein degradation via pathways that belong to the cell’s protein-degradation machinery offers new ways to reach some of the estimated 85% of undruggable targets in the proteome.
Today targeted protein degradation is achieved using several distinct pathways that involve either the lysosome or proteasome. For each pathway, step-by-step profiling of molecular events using biochemical and biophysical assays is essential to understand structure-activity relationships and to help in the rational design of new degraders.2
In this blog, we look at some of the assays and technologies available to study protein degraders. What is the preferred assay for the task at hand? How do you decide which detection technology and lab instrumentation are the best fit for your needs. There is no one answer to each of these questions but there are some guiding principles that have emerged as useful markers.
To look at assay and technology options available to study protein degraders, we will consider the detailed steps of one of the cell’s pathways for proteolysis, in this case the ubiquitin-proteasome system. The ubiquitin-proteasome system for targeted-protein degradation involves multiple steps and is the degradation pathway by which PROTACs (PROteolysis TArgeting Chimeras) and molecular glues exert their actions.
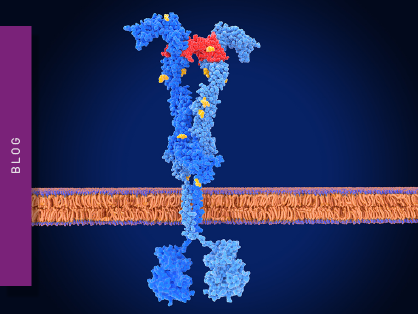
Figure 1 shows the degradation of a hypothetical disease-causing target protein via an E3 ubiquitin ligase. The pathway is often depicted as a linear sequence of events, but a circular scheme is a good reflection of what is involved since it depicts the catalytic mode of action of degraders. Namely, the target protein of interest is degraded at the end of the cycle but the PROTAC, molecular glue or other degrader is free to engage in a further round of activity.
The cycle can be divided into four main steps which include binary target engagement (activation), ternary complex formation (conjugation), target ubiquitination (ligation) and target degradation (proteolysis). The binary complex arises by binding between the degrader and the E3 ligase or the protein of interest. Next, the binary partner recruits a second partner. In the case shown in Fig.1, the E3 ligase forms the ternary complex.
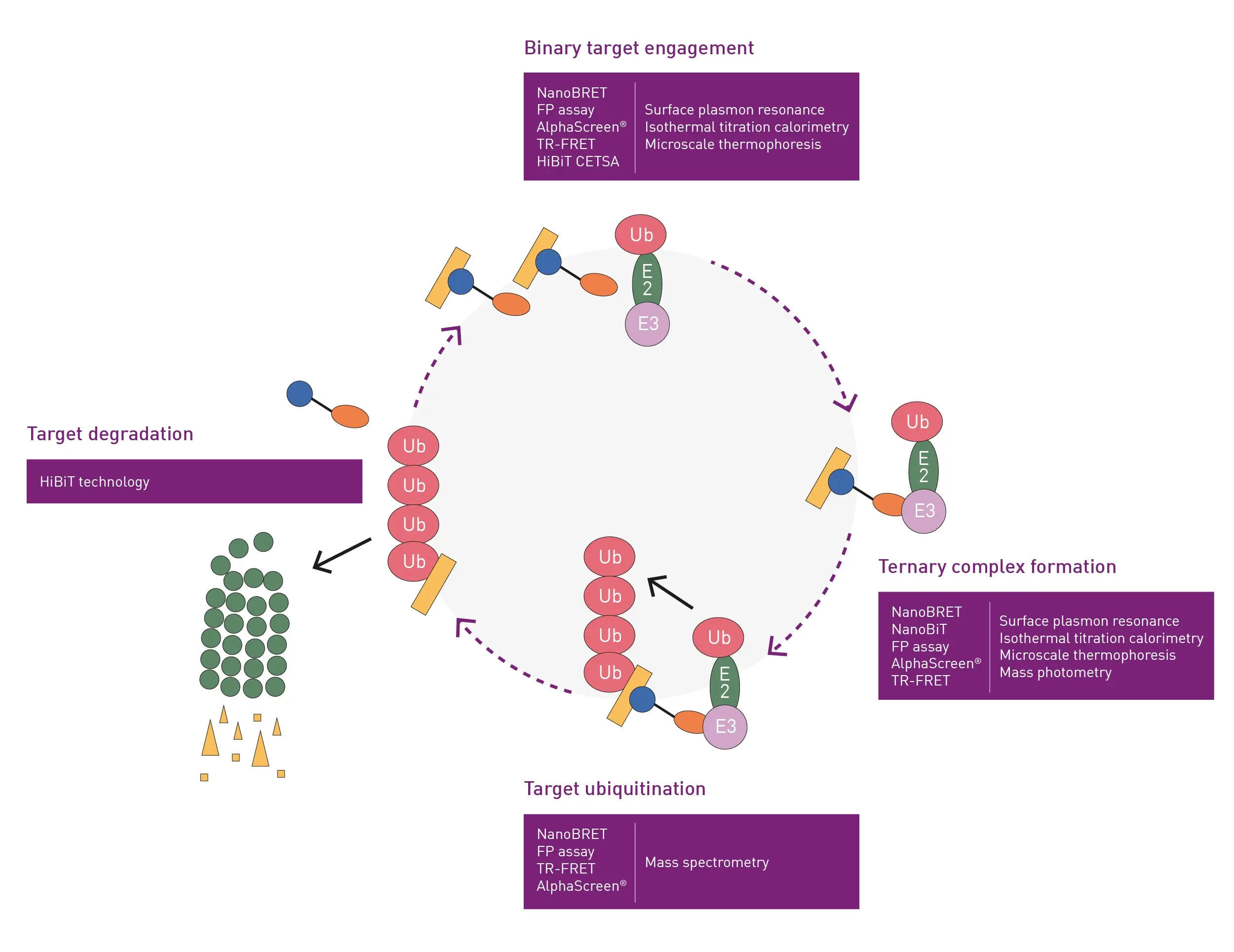 The formation of the ternary complex leads to the transfer of ubiquitin onto the substrate, polyubiquitylation and degradation of the protein of interest. As noted earlier, the degradation event leads to depletion of the degrader binding site which means that the degrader gets released back into the cytosol to undergo multiple catalytic cycles.
The formation of the ternary complex leads to the transfer of ubiquitin onto the substrate, polyubiquitylation and degradation of the protein of interest. As noted earlier, the degradation event leads to depletion of the degrader binding site which means that the degrader gets released back into the cytosol to undergo multiple catalytic cycles.
Several methods can be used to measure most of the steps shown in figure 1 for small molecule-induced degradation of proteins in vitro. For example, surface plasmon resonance, isothermal titration calorimetry and microscale thermophoresis are commonly used to measure binary complexes for protein degraders under in vitro conditions.1
Surface plasmon resonance measures changes in the refractive index in the vicinity of thin metal layers in response to protein-protein interactions or other protein interactions. It allows determination of the specificity, affinity or kinetic parameters for these types of interactions. Isothermal titration calorimetry is a label-free method for measuring the binding of any two molecules that release or absorb heat upon binding. Microscale thermophoresis also measures temperature-induced changes but in this case the motion of fluorescent molecules along a microscopic temperature gradient is detected, which reflects changes in molecular charge or size.
The same three techniques can be used to look at ternary complex formation. In addition, due to the increase in mass associated with ternary complex formation, techniques like mass photometry are also applicable for this step.
Mass spectrometry is the preferred choice to measure the increase in mass upon the transfer of ubiquitin in in vitro assays. However, it is currently not possible to measure the final degradation step itself in vitro. Overall, the instruments and devices used for measurement of these in vitro steps are heavy on resources. In addition, more than one instrument is clearly needed to assay the cascade of reactions until you reach the degradation step itself where no solution is available.
One of the biggest restrictions for in vitro assays is that the whole process from binary target engagement to target degradation is not fully compatible with high-throughput screening for these types of technologies.
A more inclusive cost-effective measurement option is to look at the complete protein degradation process in a microplate reader using live cells. For example, unlike the in vitro assays mentioned earlier, it is possible to cover the whole sequence of events in the ubiquitin-proteasome cycle using luminescence-based techniques and live cells. [As shown in Fig.1, luminescence is not the only detection option that can be chosen for each step of the pathway. The alternative detection technologies, which are shown in the figure, mainly involve different types of fluorescence and are described later in this blog.]
First, we will discuss what can be achieved using luminescence and live cell assays. Luminescence assays are often used in high-throughput screening applications due to their sensitivity and low background signal. They can be readily used to study protein-protein interactions, protein degradation or other cellular processes affected by degraders. This measurement principle is highlighted in the application note: dTAG protein degradation assay for the targeted degradation of proteins of interest where it is used for the validation of potential target proteins involved in disease.
Bioluminescence resonance energy transfer or BRET is a ratiometric luminescent-type detection method that is mainly used in cell-based assays. The donor in BRET is a luciferase enzyme that generates emission light through an enzyme reaction. Luminescence excites an acceptor fluorophore if the donor and acceptor are near one another. NanoBRET™ (Fig. 2) offers a stronger signal and less background noise compared to classically used BRET components due to modifications made to a specific luciferase enzyme isolated from a species of deep-sea shrimp. Compared to BRET, NanoBRET offers distinct advantages in the range and breadth of assays available as well as better performance due to improved spectral separation between donor and acceptor emission. NanoBRET offers an order of magnitude improvement in sensitivity over other existing BRET options. Some of the advantages of using NanoBRET over other bioluminescent assays and techniques like FRET, which include increased sensitivity, are discussed in detail in the BMG LABTECH blog article NanoBRET.

In the sequence shown in figure 1 for targeted protein degradation, the first step is a NanoBRET target engagement method where you can measure the formation and affinity of the binary complex.
For the ternary complex formation and affinity steps, a HaloTag-based NanoBRET system is a good option to probe the protein-protein interactions. A HaloTag, which is a 297-residue protein derived from the bacterial enzyme haloalkane dehalogenase, is a self-labeling protein tag designed to covalently bind to various proteins of interest. The HaloTag is composed of two covalently bound segments that include the haloalkane dehalogenase and a synthetic ligand of choice. The synthetic ligands consist of a reactive chloroalkane linker bound to a functional group which can include a selection of fluorescent dyes.
The same type of HaloTag-based NanoBRET system can be used to measure ubiquitin transfer onto the substrate (target ubiquitination).
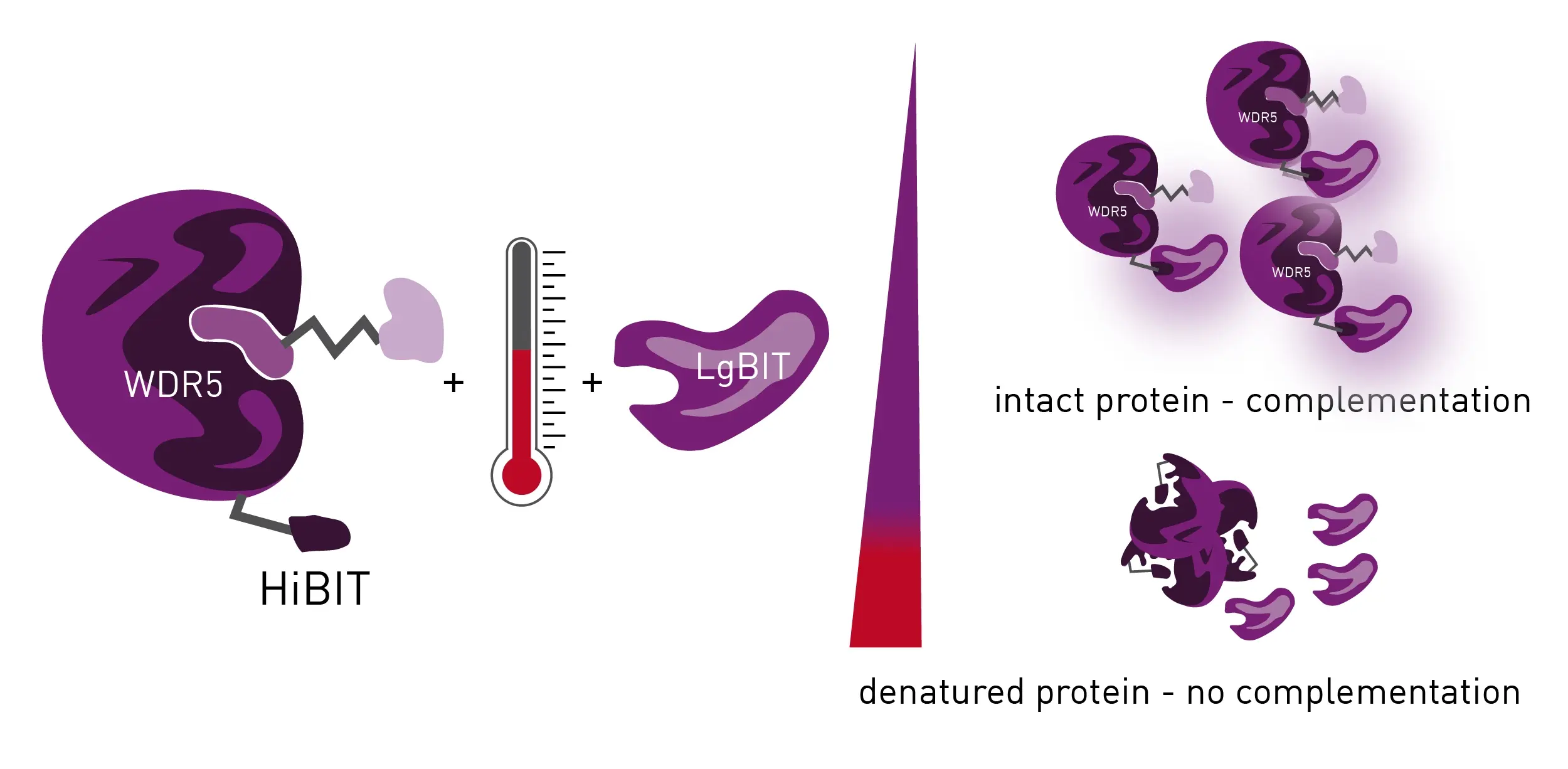
The last and most important step is to measure the actual degradation taking place in the living cells. Typically, this can be done by Western blots and selectivity screening using proteomics. However, Western blots have limited dynamic range and are not great for distinguishing between bands of similar intensity. Currently, the best way to measure protein abundance in living cells in a microplate is to use HiBiT technology (Fig.1) in a nanoluciferase complementation assay.
The HiBiT of nanoluciferase can be used to endogenously tag a protein of interest using for example CRISPR Cas9. HiBiT is a small protein (an 11 amino-acid-long beta-barrel sheet) that does not significantly change the behavior of the larger luciferase protein (LgBiT). For example, trafficking and cellular localization remain the same. When HiBiT interacts with LgBiT, luminescence takes place and the generated signal can be detected by a luminescence plate reader.
An LgBiT expression vector can be used for transfection. The LgBiT does not have to be continuously expressed but transfected into cells when needed. The protein of interest, which is labeled with the HiBiT, complements the LgBiT when expressed in the cell. Substrate is added and the signal measured at 450 nm. The treatment of cells with degrader leads to polyubiquitination and subsequent breakdown of the protein of interest. The luminescent signal therefore decreases as degradation proceeds.
Significantly, all the assays shown in Fig.1 can be run on the PHERAstar® FSX microplate reader on 1536-well microplates with a Z’ higher than 0.5.
Further information on targeted protein degrader experiments described here and the ways to evaluate molecular glues, PROTACs and other degraders are provided in this scientific talk:

One of the main challenges in degrader evaluation is often related to the size of the binder. The question often arises as to whether a binder is weak or whether it is going into the cells? Passage through the cell membrane is therefore an important parameter to measure and is needed to calculate the right dose of degrader for cells.
The strength of the interaction between a protein and a degrader can be determined using tracer molecules in indirect binding assays. Tracer molecules typically have three components: a ligand that binds to the target, a linker molecule, and a detection reagent that is often a fluorescent dye. If cells are treated with a degrader and the degrader enters the cellular system, competition takes place between the tracer and the degrader for the binding site. If the degrader binds successfully, a decrease in BRET signal can be measured and plotted against the dose of the degrader. This can be used to calculate the affinity of the unlabeled degrader to the protein target.
If a tracer molecule does not exist and is not an option, a tracer-free assay system like a HiBiT thermal shift assay (BiTSA) can help. The application note Measuring protein ligand binding with an endogenous HiBiT CETSA® test system describes such an assay and how it can be used to screen for PROTAC and protein of interest (POI) interactions (Fig. 3). The principle is based on the thermal stabilization of a protein that occurs when a compound binds to a protein of interest. The bound state of such an interaction demonstrates more stability compared to the non-bound state. Thermal stabilization can help rank compounds according to affinity but does not give IC50 values. Melting curves can be generated that show the highest and lowest binders but affinity values cannot be calculated.
You can learn more about the nature and significance of tracers at tracerDB, a resource with more than 300 proteins accompanied by more than 40 tracers in experimentally validated assays.3
Investigators have highlighted the importance of characterizing the ternary complex to guide the design of protein degraders.4 Mechanistically, a lot depends on the rate limiting formation of the ternary complex which correlates with the final degradation event. It thus makes a lot of sense to screen for ternary complexes rather than binary complexes for targeted protein degradation. The ternary complex correlates more closely to the final degradation step in the pathway.
Kinetic measurements are important when looking at degraders. Some proteins of interest are highly responsive upon initial degrader treatment. The targeted protein can disappear in a few hours. Resynthesis rates can be so high that degradation can be easily missed using techniques like Western blots if the sampling times are not at the right time intervals. The use of kinetic assays on microplates can help avoid this type of challenge. Degrader activity can be represented by DC50 and Dmax values. The DC50 value is the concentration at which half of the target protein is degraded and is a measure of the potency of the degrader. Curves at different concentrations of degraders can be used to determine DC50 values. These values are useful indicators of what concentrations of specific degraders to use in different experiments.
In addition to luminescence-based methods, different types of fluorescence technologies are well suited to look at the different steps involved in targeted protein degradation. However, it should be noted that these fluorescence technologies are mainly used in a biochemical setting that involves purified proteins in solution or cell lysates. Unlike luminescence-based approaches, these methods are not really suited for studies in live cells. For instance, donor lanthanide or AlphaScreen beads are excluded from transfer into cells due to their size and structure. Accordingly, luminescence remains the first choice for live cell-based degradation analyses.
Some of the additional options available are as follows:
Fluorescence intensity measurements are often used in reporter assays. Under the right conditions, fluorescence intensity correlates linearly with the concentration of the molecule to be measured (e.g., analyte coupled to the excited fluorophore) which makes it appropriate for quantitative analyses.
FRET and TR-FRET are often used to study molecular interactions or binding events. FRET and TR-FRET both depend on energy transfer between two fluorophores. In the case of TR-FRET, the donor fluorophore is a longer-lived lanthanide which results in significant improvements to background noise levels. TR-FRET (Fig. 4) offers significantly reduced background and an increased detection window between negative and positive signals compared to FRET. The lanthanide emission is more stable over time. As a homogeneous assay, detection of the donor-acceptor complex does not require physical separation from unbound components. This eliminates the need for separation steps as well as the multiple washing steps needed in other methods. An example of this is highlighted in the application note: Studying intramolecular bivalent glues using TR-FRET binding assays.
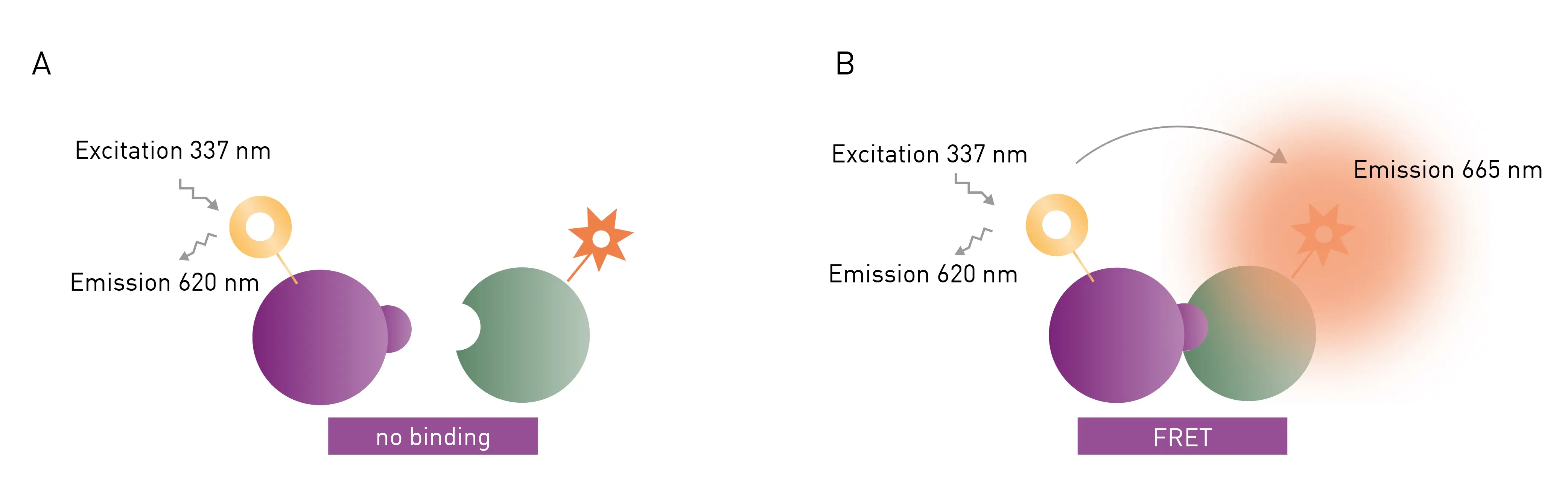
Fluorescence polarization (FP) depends on measurement of the parallel and perpendicular emission intensities of plane polarized light. Polarized light arises when the direction of the electric field of light is restricted to one plane by a specific filter. In this case all selected light waves oscillate in a single direction. FP is widely used to study molecular interactions in solution, including protein-protein interactions, protein-DNA binding, enzyme activity and other molecular interactions.
In the application note High-throughput screening and hit validation with a fluorescence polarization assay an FP assay was developed based on the use of a fluorescently labeled peptide ligand that binds to LC3A. LC3A is a member of the human Atg8 protein family and is only found in the autophagosome. The discovery of potent LC3A inhibitors serves as a handle for the development of ATTECs (AuTophagy-TEthering Compounds) and offers the possibility for novel targeted protein degradation approaches.5 ATTECs are a novel class of bifunctional molecules proposed to hijack the autophagosomal pathway for the degradation of cellular components including proteins. An ATTEC helps promote fusion of the autophagasome with the lysosome enabling the two structures to work together to degrade the target protein.
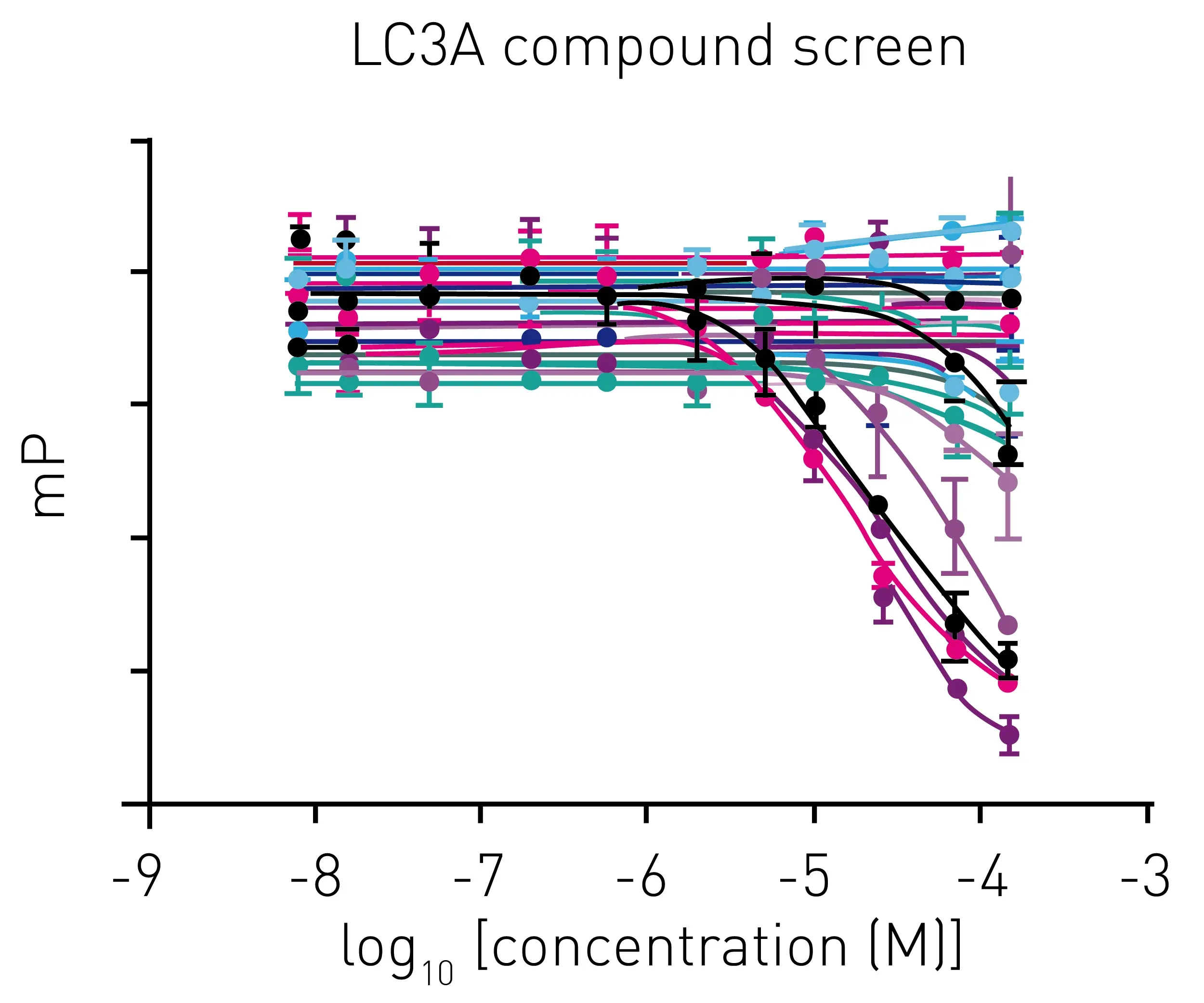
In the study, p62 LIR peptide, a peptide ligand of LC3A, was labelled with a Cy5 fluorophore to act as a tracer molecule. The addition of competing ligands of LC3A to the LC3A-tracer complex results in a displacement of tracer and a reduction of the fluorescence polarization signal (Fig.5)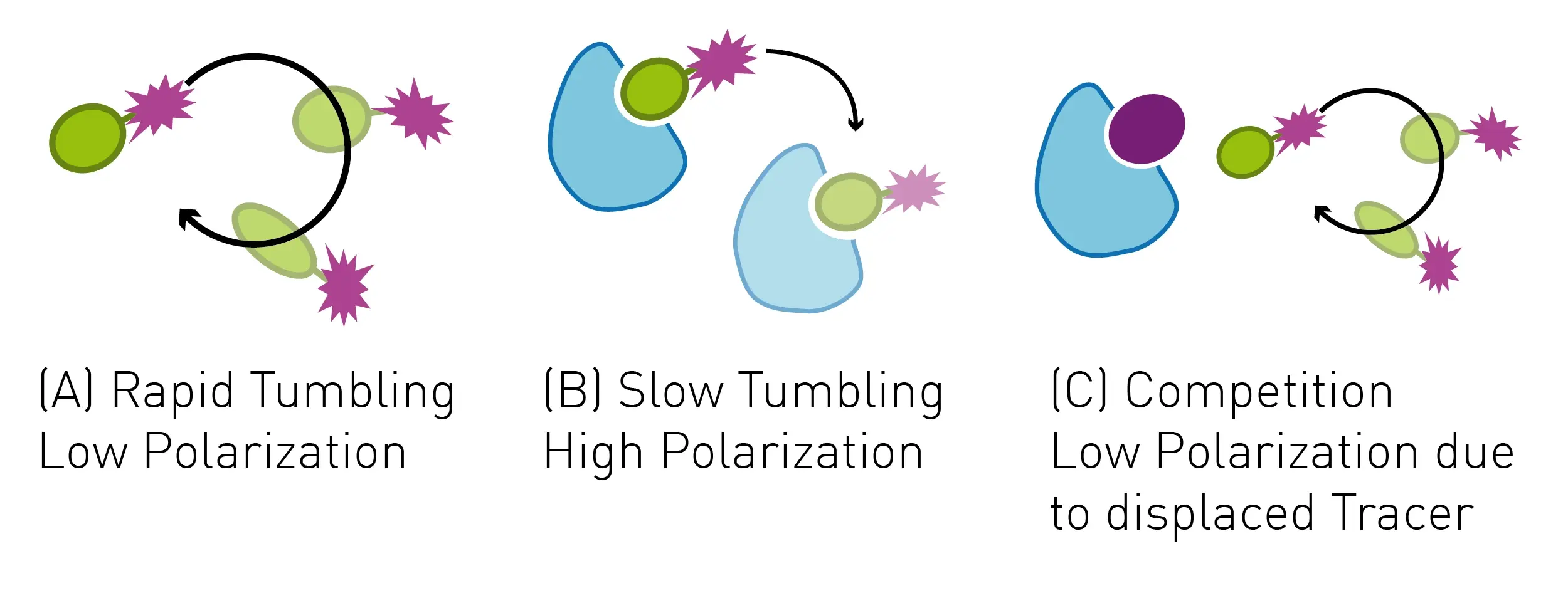
Miniaturization of this assay allowed for library screening in 1536-well plates (Fig. 6) that was suitable with automation for screening of large compound libraries in days.
 Fluorescence polarization assays can also be used to study the binding activity of molecular glues to their targets and the kinetics of interactions. Changes in fluorescence polarization due to fluorescently labeled molecular glues binding to target proteins may be measured. This can provide information about the binding strength and kinetics of the interaction between a target protein and degrader.
Fluorescence polarization assays can also be used to study the binding activity of molecular glues to their targets and the kinetics of interactions. Changes in fluorescence polarization due to fluorescently labeled molecular glues binding to target proteins may be measured. This can provide information about the binding strength and kinetics of the interaction between a target protein and degrader.
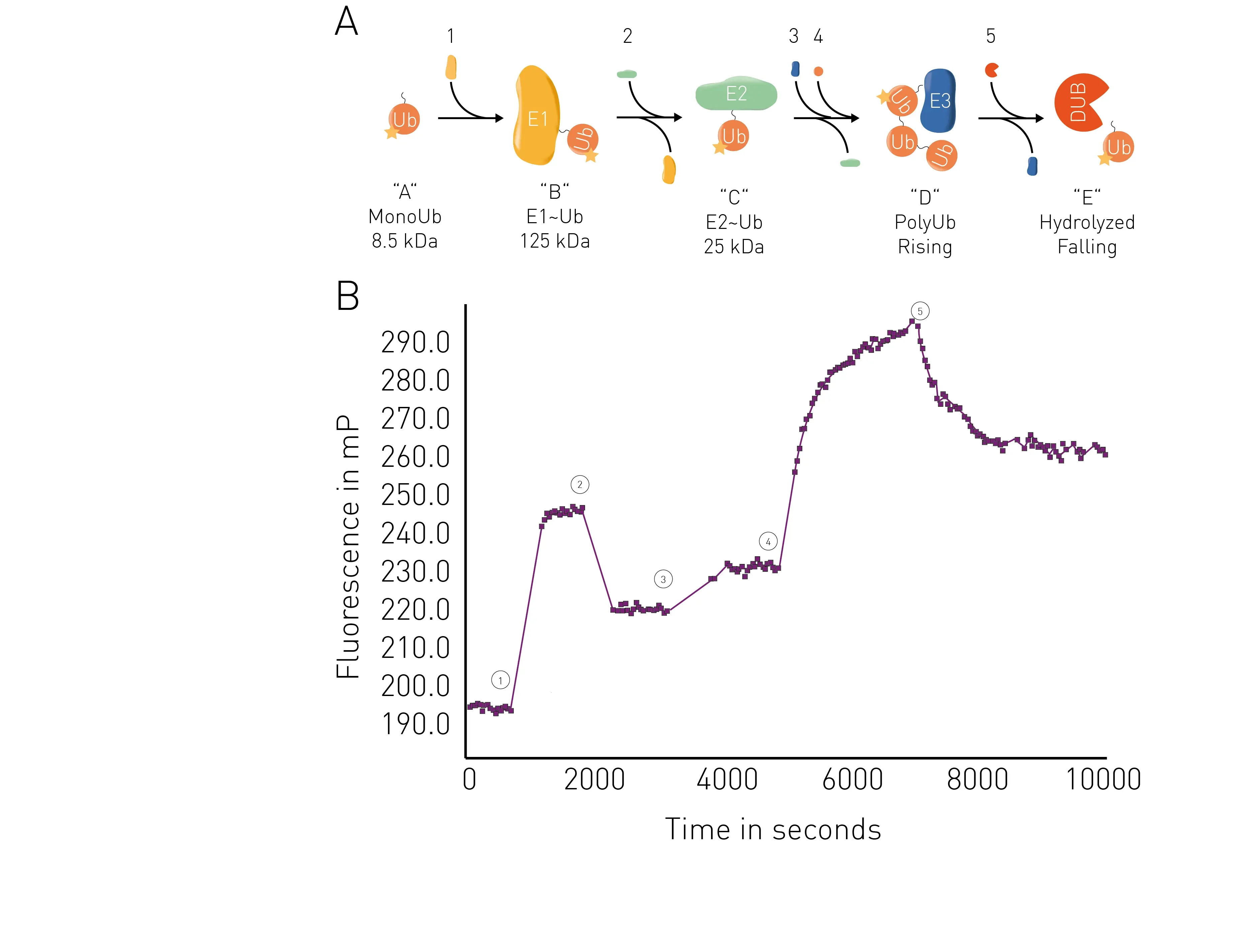
The application note Ubiquitination monitoring in real-time: the fluorescence polarization-based method UbiReal describes a fluorescence polarization method that can be used to track all stages of ubiquitin conjugation and deconjugation in real time. The approach is suitable for high throughput formats and fluorescence polarization is used to measure fluorescently labeled ubiquitin. All stages of the ubiquitination cycle can be monitored using UbiReal (Fig.7).
AlphaScreen® is a family of related technologies often used for high-throughput screening in the life sciences. Amplified Luminescent Proximity Homogeneous Assays like AlphaScreen offer bead-based chemistry that can be used to study interactions between molecules in a microplate or used as immunoassays to quantify analytes. At the heart of the AlphaScreen assay is the diffusion of singlet state oxygen from donor to acceptor beads. When a biomolecular interaction takes place that brings the donor and acceptor beads into proximity, a cascade of chemical reactions ensues that amplifies the luminescent signal in the region 520-620 nm.
Again, as mentioned earlier, the fluorescence technologies mentioned above are mainly used in a biochemical setting. Unlike luminescence-based approaches, these methods are not really suited for studies in live cells. Accordingly, luminescence remains the first choice for live cell-based degradation analyses.
Microplate readers provide distinct features that support scientists looking to perform research on targeted protein degraders.
High throughput screening enables researchers to quickly evaluate and run diverse assays that support the needs of drug discovery and development of degraders.
The PHERAstar® FSX delivers measurements at high sensitivity and with the detection times needed for automated analysis. The ability to use high-density microplate formats such as 384-, 1536- or even 3456-well microplates means low sample volumes translate into cost-effective solutions.
The Simultaneous Dual Emission (SDE) feature also significantly enhances read time and throughput for accurate measurements of samples in high-density formats. Detection technologies like TR-FRET and BRET, for example, emit at two wavelengths and the ratio of the measurements at these two wavelengths is reported. In fluorescence polarization, SDE can be used to read both the parallel and perpendicular emission signals at the same time. The PHERAstar® FSX is equipped with two paired sets of photomultiplier detectors that simultaneously monitor both emission wavelengths from a FRET, BRET and FP or TR-FRET assay. SDE means that data can be collected in one detection pass rather than two, which halves read times and doubles the throughput.
Cross-talk is interference caused by stray light from adjacent wells that may artificially impact the signal in a specific well of a microplate. When running glow luminescence and AlphaScreen® assays in high-density microplate formats, cross-talk can negatively affect data quality and statistics (Fig. 8). Solutions for cross-talk reduction improve data quality especially in screening assays. Hardware- and software-based solutions for cross-talk reduction are built into plate readers like the CLARIOstar Plus, VANTAstar®, and PHERAstar FSX. 
As mentioned earlier, even though binding kinetics for targeted protein degradation can be analysed by technologies like surface plasmon resonance, isothermal titration calorimetry or microscale thermophoresis, microplate readers offer a way to look at the complete sequence of protein degradation events relevant to degraders in an efficient, cost-effective manner. Binding kinetics can be miniaturized into a microplate format, which enables the high-throughput kinetic screening of large compound libraries. In particular, the PHERAstar FSX offers unique and dedicated features to efficiently monitor kinetic binding events using FRET, TR-FRET, FP and BRET kinetic detection.
If you are relying on TR-FRET interaction assays to analyse kinetic binding, the PHERAstar® FSX plate reader can be equipped with a TRF laser. Besides providing wavelength-specific high energy excitation, the TRF laser allows for faster detection (60 Hertz flash frequency) at high sensitivity and with optimal statistical performance.
As we saw earlier, the ability to perform cell-based assays is an asset for research into targeted protein degraders. Bottom reading capabilities and atmospheric control are both useful in this context.
Bottom reading capabilities provide robust measurements for cell-based assays since the distance between the adherent cell layer and detector is reduced. This minimizes unwanted effects including those from the cell culture medium. Microplate readers offering a software-controlled top/bottom detection switch provide flexibility and avoid the need for manual intervention, which is particularly important for automation.
A physiological environment is particularly beneficial when running cell-based kinetic assays. BMG LABTECH´s Atmospheric Control Unit (ACU) provides an optimal environment for real-time cell-based assays where stable O2 and CO2 tensions can be maintained.
Knowledge of PROTACs and molecular glues has been built over decades. New technologies for targeted protein degraders are emerging at pace along with approaches targeting different degradation routes and even ways to degrade larger components of cells including defective organelles. The field has come a long way since the Nobel Prize in Chemistry was awarded to Aaron Ciechanover, Avrim Hershko and Irwin Rose in 2004 for the discovery of regulated protein degradation. Not many people were interested in how proteins were broken down at that time. But the discovery of ubiquitin tagging of proteins and their cyclical degradation in the proteasome not only improved understanding of how processes like cell division, DNA repair, quality control of proteins and immunology are governed but also strengthened the idea that useful drugs could arise from looking at the native degradation systems of the cell.
BMG LABTECH is ideally placed to support existing and future needs in this promising area of research that offers new opportunities to reach previously undruggable protein targets.
What is the preferred BMG LABTECH microplate reader for specific needs and applications related to molecular glue degrader research? BMG LABTECH offers a range of detection devices for sensitive absorbance, fluorescence and luminescence measurements.
The PHERAstar FSX was specifically conceived for screening campaigns and is your go-to reader for high-performance high-throughput investigations in molecular glue research.
Both the VANTAstar® and CLARIOstar Plus allow for wavelength scanning and include Enhanced Dynamic Range technology for superior performance in a single run. They also offer increased light transmission and sensitivity courtesy of Linear Variable Filter MonochromatorsTM and different filter options. In addition, they can be equipped with the Atmospheric Control Unit for live cell-based assays.
All BMG LABTECH microplate readers have exceptionally fast reading capabilities. In addition, the Omega series, CLARIOstar Plus and PHERAstar FSX microplate readers come with on-board injectors that can offer the very best options for detection at the time of injection.
Collectively, these multi-mode readers combine high performance with miniaturized assays, short measurement times, and offer considerable savings on materials and other resources.
Configure your microplate reader and get an initial recommendation!
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Flexible microplate reader with simplified workflows
Upgradeable single and multi-mode microplate reader series
Binding constants quantify the strength of a binding reaction between a biomolecule and its target (ligand). But how do you measure them and what can you do with them?
Receptor-ligand kinetics is the study of the rates at which receptors and ligands interact, bind and dissociate. Learn why these types of measurements are important and how to measure them.
Receptor-ligand interactions are crucial for cell signalling. They are also important for drug discovery. How do microplate readers deliver benefits to both?
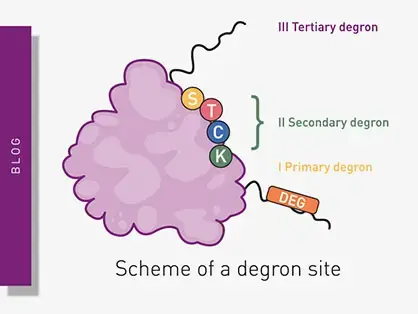
Degrons are specific sequences of amino acids or structural motifs within a protein that are important for targeted protein degradation. Find out how microplate readers can advance research into natural and engineered degrons.
Nitration can significantly modify the structure and function of proteins in cells. But how can measurements of protein nitration reveal crucial information about biological activity and how can microplate readers help?
Innovation for targeted protein degradation and next-generation degraders is gathering pace. This blog introduces some of the different approaches that act via the lysosome or proteasome.