
CLARIOstar Plus
Most flexible Plate Reader for Assay Development

Fluorescence is based on photoluminescence, a process of glow and light emission. It is a physical process in which light is emitted after it has been absorbed by a substance. The fluorescence intensity indicates how much light (photons) is emitted. It is the extent of emission and it depends on the concentration of the excited fluorophore.
Fluorescence is created by the absorption of energy (light) by fluorescent molecules, called fluorophores. The absorbed energy raises electrons to a higher energy level. As this excited state is unstable, the electrons fall back to the ground state and thereby emit light (fig. 1).
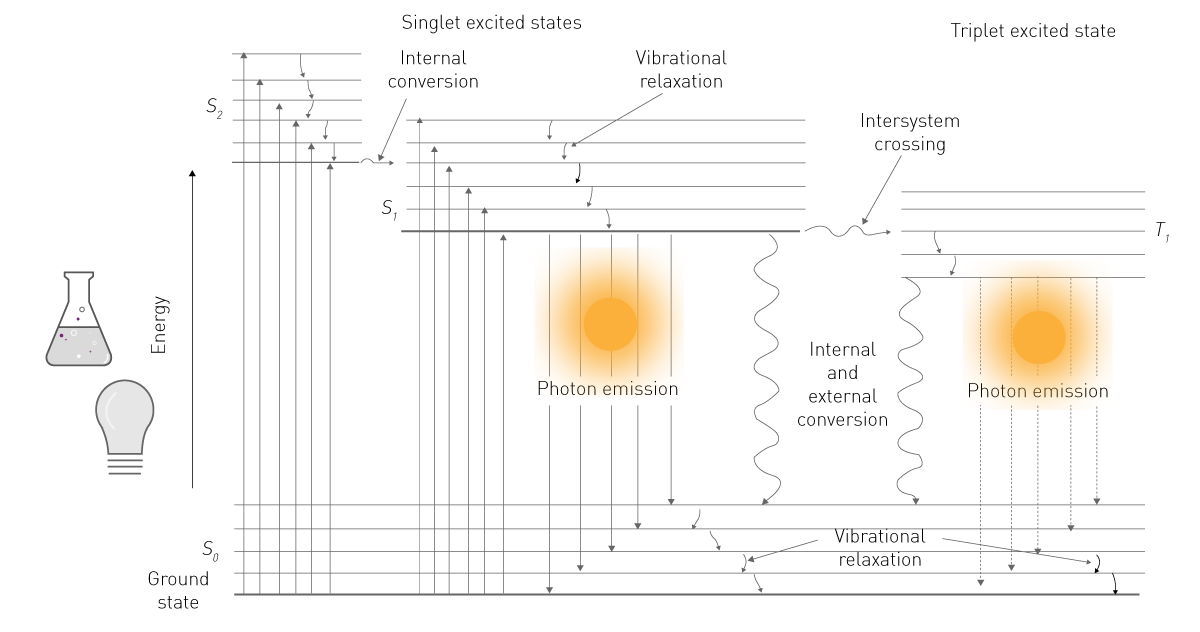
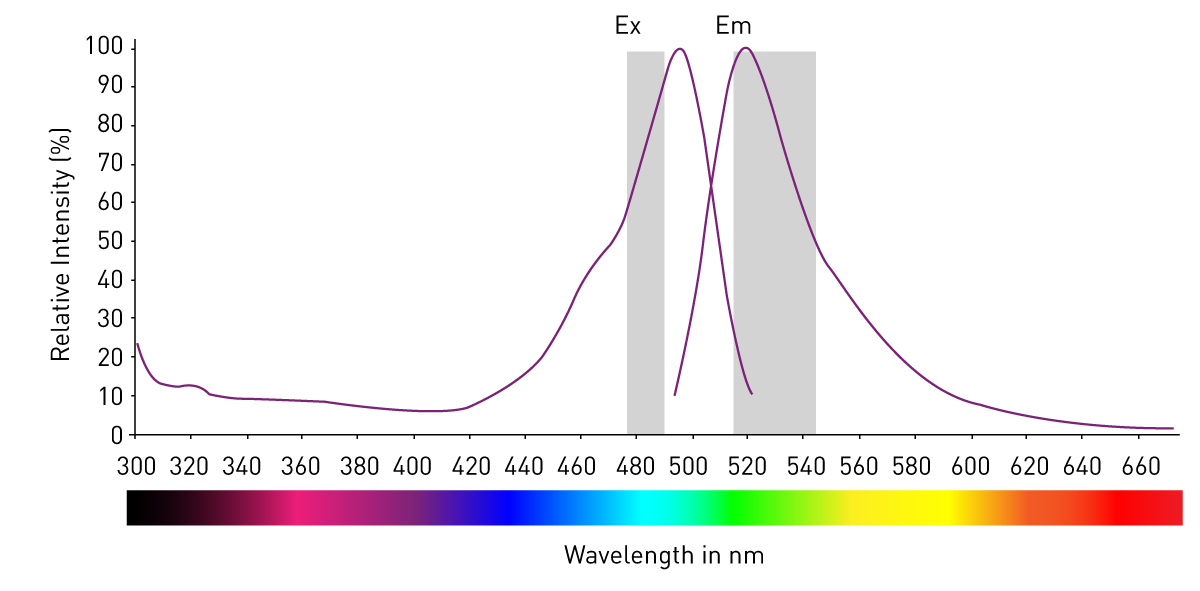
A smaller part of the absorbed photons is converted into movement and heat. Thus, the energy that is released when the electrons fall back is always smaller than the energy required to excite the electron. Since higher-wave radiation is less energetic than short-wave radiation, the emission has always a higher wavelength than the light that was used for excitation. The distance between excitation and emission maxima (peaks) is called the Stokes shift (fig. 2).1
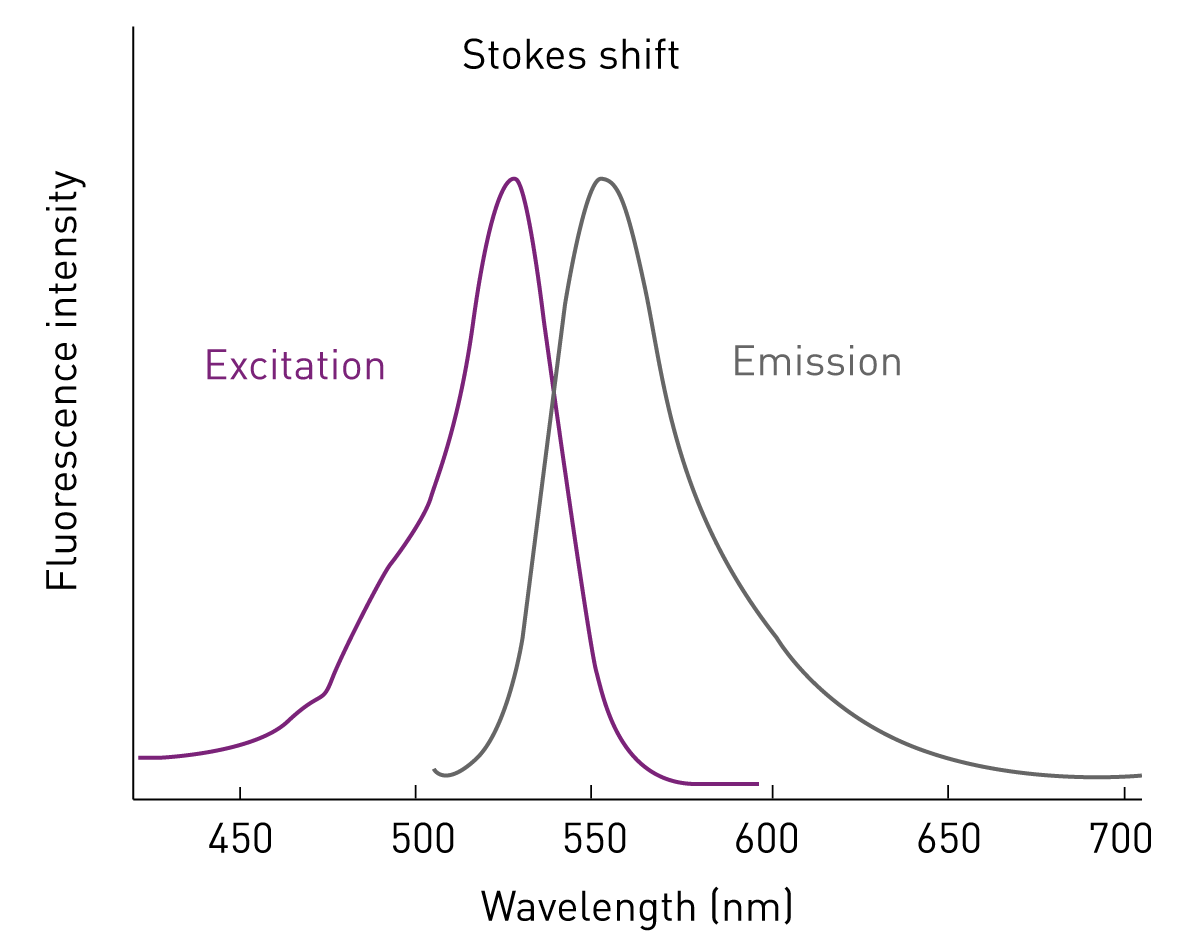
The characteristic of the excitation (absorption) light being at a lower wavelength than the emission light opens the possibility to use fluorescence intensity for analytical purposes. Fluorophores can be detected by exciting them at a specific wavelength and measuring their emission intensity at a higher wavelength. Fluorescence intensity is linearly correlated to the concentration of the excited fluorophore and is consequently suited for quantitative analyses.
Fluorescence intensity is widely used in life science applications: in microscopy to localize and quantify biomolecules, in flow-cytometry to analyze cells, and in microplate-based assays to quantify molecules, enzymatic activities, and even interaction between molecules. This page focuses on the use of fluorescence intensity in microplate assays, how it is detected, what to consider for fluorescence intensity measurements, and which assays are commonly used.
A fluorophore (also fluorochrome) is a fluorescent compound that can emit light upon excitation by light. Thousands of molecules are known that exhibit fluorescent characteristics.2 They differ in:
Many fluorophores are chemical compounds with a small molecular size that can be easily linked to other molecules. Fluorescent proteins such as green or red fluorescent proteins (GFP or RFP) are larger in size and can be expressed by cells. They are used to tag proteins for expression studies, for endogenously expressed biosensors, or simply as cell quantification markers. Read how transfection efficiency based on GFP and mcherry can be monitored accurately with the VANTAstar here.
Light excitation and emission characteristics of a fluorophore are displayed as spectra: in a coordinate system, nanometers are indicated on the x-axis and intensity on the y-axis. The curves are normalized to 100% and they allow quick identification of maximum excitation and emission wavelengths as well as of the Stokes shift. This information is important to choose the right fluorophore for an assay. It needs to be compatible with the detection system, potential autofluorescence, and other fluorophores if several are used in an experiment.
The quantum yield is a measure of how efficiently an excited molecule converts absorbed photons into emission light. In combination with the extent to which light is absorbed (extinction coefficient), the quantum yield gives an indication of the brightness of a fluorophore.
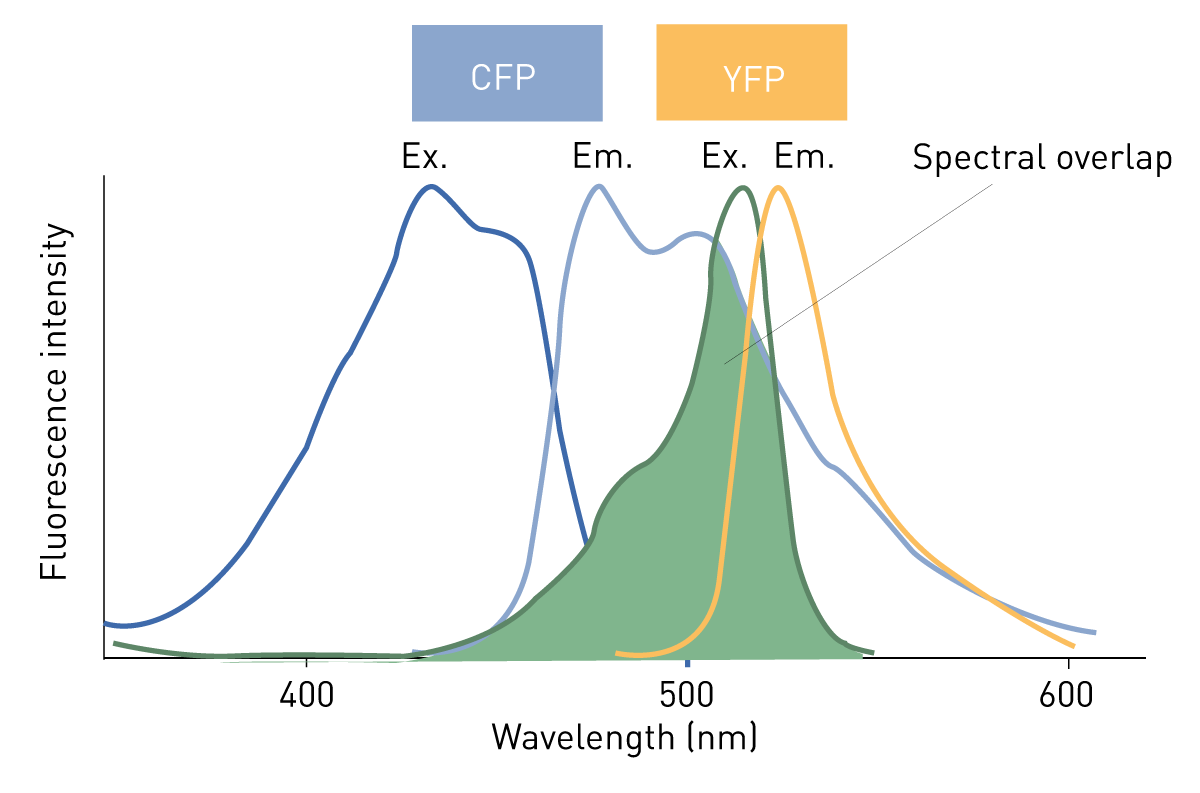
FRET (Förster´s Resonance Energy Transfer) describes the energy transfer from a donor fluorophore to an acceptor fluorophore. The transfer happens when the acceptor gets excited by the emission light coming from the donor and only takes place if both fluorophores are in proximity. FRET is often used for molecular binding studies like receptor-ligand interactions or in biosensors. The latter change their conformation when the analyte binds and brings the two fluorophores together. For FRET to take place, the fluorophore pair needs to have an overlap between the donor emission spectrum and the acceptor excitation spectrum.3
Even though fluorophores differ from each other, the basis of fluorescence intensity measurements in microplates is the same (fig. 4):
As opposed to absorbance, fluorescence is not an absolute measurement. The intensity of the fluorescent signal is usually relative to other measurements or to a refence measurement taken by an instrument. Consequently, fluorescence plate readers measure the light signal emitted by a sample in Relative Fluorescent Units (RFU).
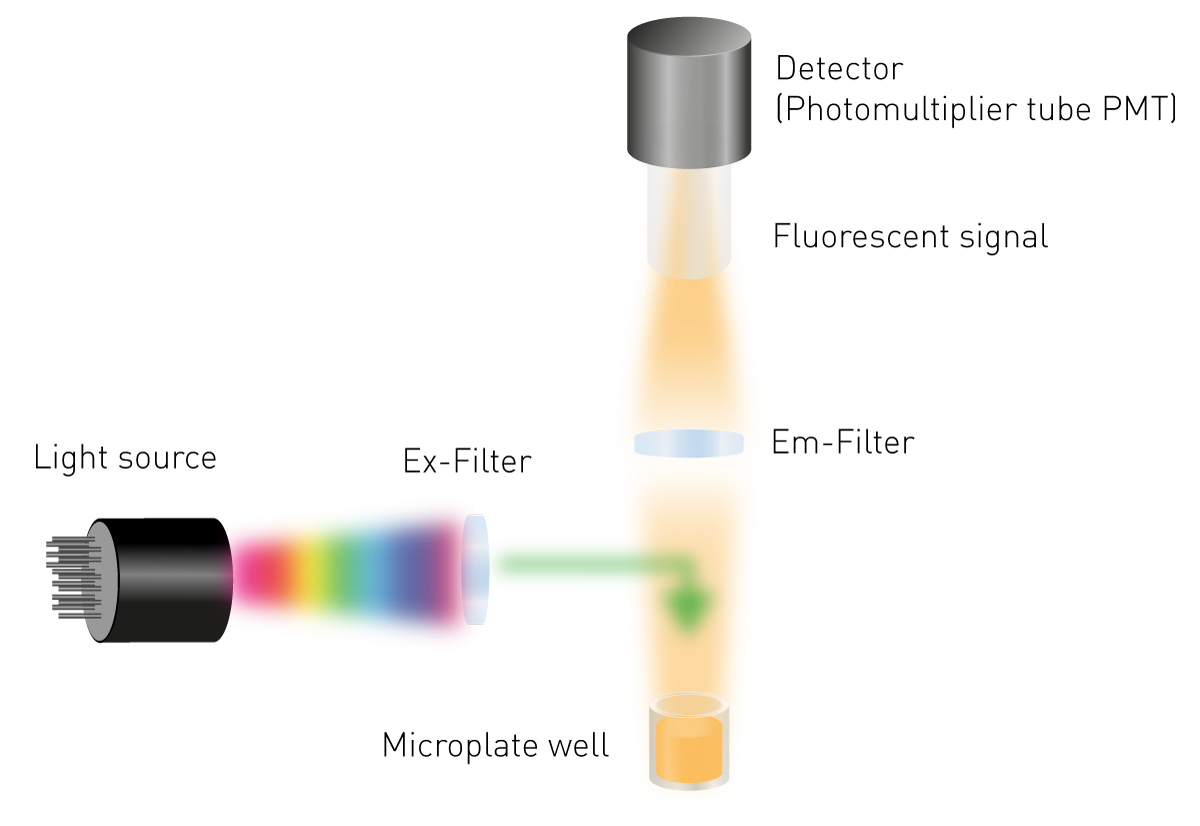
There are three commonly used light sources useds in microplate readers. Regardless of which light source your reader is equipped with, its performance can affect measurement results. Read more in the HowTo Note: How does the number of flashes influence measurement results?
Xenon flash lamps emit light in the range of 200 nm to 2500 nm. Therefore, they are suited to excite any molecule that absorbs light from the UV range to the infrared range. They exhibit a high signal output and are long-lived. Because of these advantages, xenon flash lamps are used as fluorescence excitation sources in BMG LABTECH plate readers.
Halogen lamps emit light starting from approximately 360 nm and are unsuited to measure UV fluorescence intensity. In comparison xenon lamps, tungsten lamps have a shorter lifetime, a lower output intensity, and result in lower sensitivity, if used in microplate reader fluorescence detection.
Light Emitting Diodes (LEDs) emit light with high intensity but only at a specific bandwidth. Accordingly, multiple diodes are required for different excitation wavelengths, making flexible detection of multiple fluorophores more complicated and expensive.
Broadband excitation sources can cover wide wavelength ranges starting from UV up to infrared light. In order to specifically excite the fluorophore of interest and to avoid unspecific signals, the broadband excitation light needs to be selected (filtered) to transmit only light around the excitation maximum of the fluorophore. Similarly, the emission light is filtered for the emission maximum of the fluorophore. This way, only the fluorescence coming from the fluorophore of interest is guided to the detector. Accordingly, assay sensitivity is enhanced by optimising the wavelength range.
The sensitivity of detection in some fluorescence intensity microplate readers is enhanced by the use of an additional selection between excitation and emission: a dichroic mirror. The dichroic mirror typically transmits light higher than a specific wavelength but blocks light at lower wavelengths. The cut-off lies between excitation and emission and provides lower leak-through of excitation light to the detector.
Optical filters are thin round or rectangular glass slides with an optic coating. The coating only transmits light of a certain wavelength. Fluorophores with different excitation and emission maxima need different filters. A filter is typically named after the middle transmission wavelength, which is often the maximum of excitation or emission of the fluorophore. Furthermore, an optical filter has a defined bandwidth (or bandpass), this states how broad the transmission range is. For example, a 485 nm filter with a bandwidth of 10 nm filters white light in the range between 480 and 490 nm.

In contrast to optical filters, monochromators are capable of selecting light of different wavelengths depending on the requirement. Two types of monochromators are used in microplate readers: conventional grating-based monochromators or linear variable filter monochromators. In grating-based monochromators, wavelengths are selected by diffraction of white light into its colors. Wavelengths are then selected by a slit, which physically blocks unwanted light and transmits only the desired one. Disadvantage of the grating method is low light transmission and the occurrence of stray light, which might reach the detector and lowers overall fluorescence sensitivity.
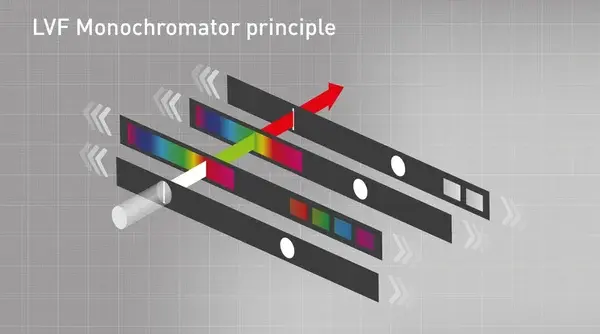
The Linear Variable Filter (LVF) Monochromator overcomes the straylight problem as it does not break up the light, but literally filters it. The LVF Monochromator uses two slides with a gradient coating. Any wavelength with any bandwidth can be selected by sliding the two linear filters against each other. The LVF Monochromator has a filter-like transmission, eliminates straylight, and thus has a better performance than diffraction monochromators (fig. 6). The use of Linear Variable Filters in microplate readers is a patented technology by BMG LABTECH.
Fluorescence intensity in microplate readers is detected with photomultiplier tubes (PMTs). They multiply the signal using the photoelectric effect and convert the light into an electric signal. For fluorescence intensity measurements in microplate readers, two different PMTs are used, which differ in their sensitive wavelength range. A low-noise blue/green PMT is sensitive in the range up to 740 nm and is often also used for luminescence measurements. A red-shifted PMT measures the fluorescence sensitively up to 900 nm and thus also covers red and infrared dyes. These are often used for cell-based measurements since the autofluorescence is reduced in this wavelength range.
Fluorescence intensity quantifications require a black plate as it reduces the background and reflection of the excitation light. More details on the plate choice are found in our blog post "The microplate: utility in practice."
Choosing the right filter is essential for a precise fluorescence intensity determination. As already mentioned, the mean wavelengths should be at or close to the excitation and emission maximum. However, there are a few things to keep in mind:
If the edges of excitation and emission filters are too close to each other, the excitation light may leak through and reach the detector. Then the signal of the fluorophore disappears in the excitation light and is no longer measurable. How close the filters can be, depends on the quality of the filters and on the use of a dichroic mirror. Typically, the highest transmitted excitation and the lowest transmitted emission wavelengths should be 30 nm apart (fig. 7).
The bandpass of filters varies from a few nanometers up to 100 nm. The bandpass affects the amount of light that can pass the filter. Low bandwidth filters (~10nm) are recommended for bright fluorophores or for complex samples with high autofluorescence. A typical example is green fluorophores such as AlexaFluor488, FITC, or GFP in a cellular environment.
Broader bandwidth filters are recommended for low emission fluorophores if they emit in a range where autofluorescence is low (emission <600nm) or if the buffer contains no other fluorescent compound. BMG LABTECH selects the filters based on the fluorophore excitation and emission spectra. In addition, filter pairs are measured and matched to one another so that you can be sure that you are receiving the best possible filters. Default settings for common fluorophores are also already stored for the monochromator so that this selection work is not left to the researcher.
The above considerations also apply to FRET measurements. However, it must be noted that two emissions must be recorded separately. The excited donor fluorophore emits light at a specific wavelength if no acceptor is nearby. If an acceptor fluorophore is close, the donor transfers the energy to the acceptor which then emits light at a higher wavelength. This means that two emission filters are required, one of which only allows the donor emission and the other only the acceptor emission to be transmitted.
The fluorescence gain is an amplification factor, or more precisely, the voltage of the detector at which the incoming light signal is amplified. Changing the gain moves the dynamic range of detection along the concentration curve of the analyte/assay. However, the width of the dynamic range is fixed and does not change.
Weak signals need a large amplification (high gain), and bright signals need less amplification (small gain). An incorrectly selected gain can make measured data unusable because it either exceeds the measuring range or differences are no longer resolved. Data highlighting these effects can be found in the HowTo Note: How to optimise the gain setting of your microplate reader? The gain is typically set to have a maximum measurement output on the sample with the expected highest intensity. This is done in order to have the largest possible dynamic window between the highest and the lowest measurement values (fig. 8).
All BMG LABTECH microplate readers determine the gain automatically, provided a specific well with a specific signal intensity is identified. However, on the CLARIOstar Plus and VANTAstar, no gain adjustment is necessary. The Enhanced Dynamic Range (EDR) technology was specifically designed to offer the largest possible dynamic range - 8 concentration decades. This provides researchers with an unprecedented convenience in microplate measurements as highly reliable results can be measured over a large dynamic range with no manual intervention.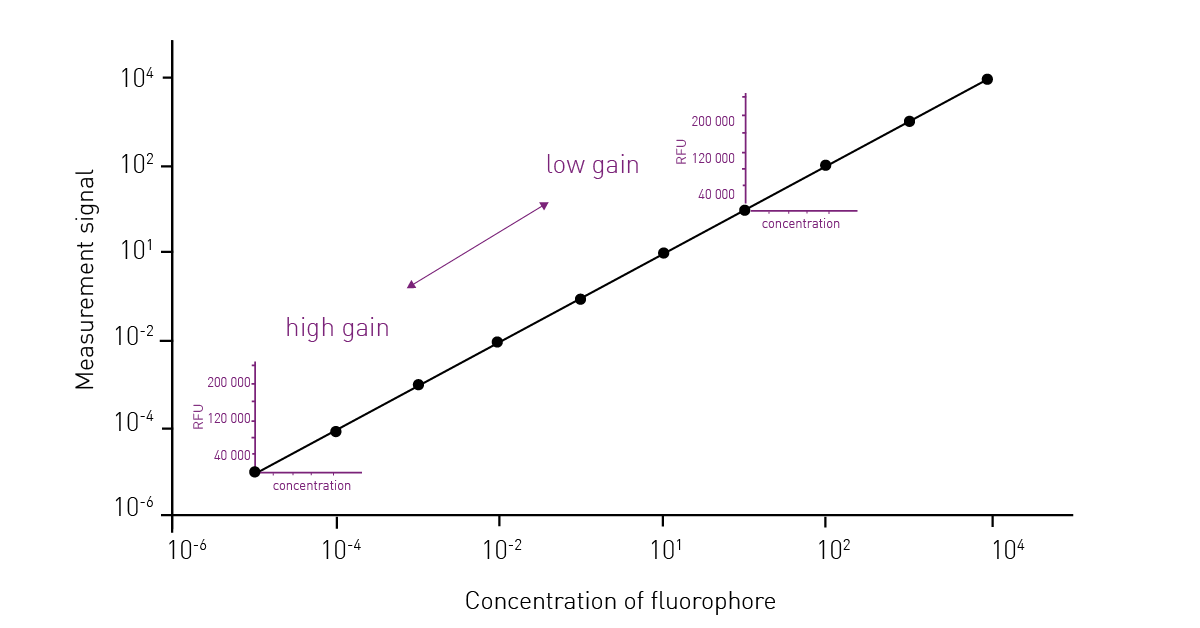
The height at which a signal is detected has a significant impact on the sensitivity of the measurement. Measuring at a not optimal focal height may reduce the signal by up to 90 %. For instance, adherent fluorescent cells need to be measured close to the plate bottom, whereas a homogenous fluorescent solution gives the highest signal just beneath the liquid surface and depends on the fill volume of the well. Some plate readers determine the optimal focal height in a selected well. The CLARIOstar® Plus and VANTAstar® go even further and determine the focal height automatically while reading a plate.
The number of fluorescent assays is as big as the number of biological and chemical questions they can answer. Therefore, the most common applications based on fluorescence intensity are outlined below.
Whether cells are harmed by a compound or cancer cells die upon treatment are common questions answered by fluorescent viability and cytotoxicity assays and microplate readers. In the alamarBlue™ assay, resazurin is reduced by metabolically active cells to a fluorophore and reports on cell viability. Another way to assess cell viability is the incorporation of fluorescently marked nucleosides into the DNA. These are incorporated and consequently fluoresce only if DNA replication takes place, a marker of viability.
A way to detect cell death by fluorescence intensity is based on fluorescent DNA dyes such as SYBR® Green or Celltox™ Green. The dyes are non-cell-permeable but get exposed to DNA in solution in case the cell membrane is disrupted during cell death and then starts to fluoresce (fig. 9). These assays are compatible with real-time measurements and can monitor cell health over time. As these measurements are done over days, the microplate chamber needs to be incubated at 37 °C and with 5 % CO2 to keep cells happy. The CLARIOstar Plus with Atmospheric Control Unit has proven to provide the ideal atmosphere for long-term cell-based experiments. More cell health assays are described in our blog post “Cell viability assays – Measure how happy your cells are”.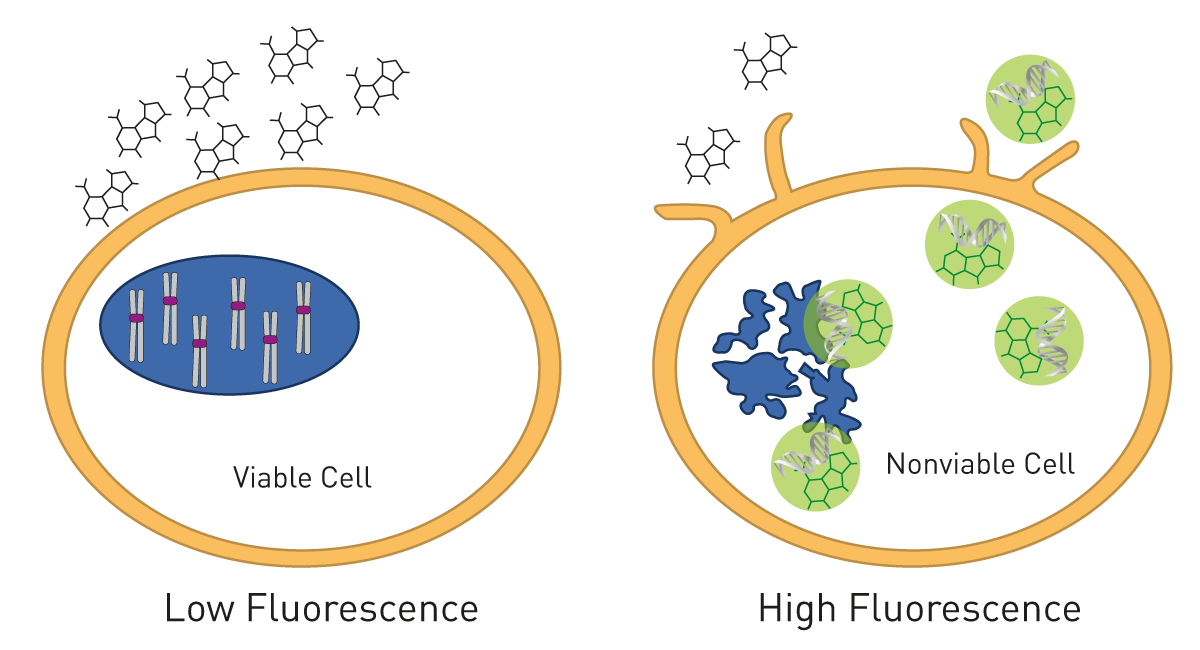
As enzymes are involved in multiple biological processes, it is important to study how inhibitors or promoters influence enzyme activities. The principles of enzyme assays are comparable: they employ a pre-fluorescent substrate which upon enzyme processing releases a fluorophore. Common fluorophores are based on coumarin derivates such as in the fluorescence-based assay of the epigenetic enzyme histone deacetylase 1 (HDAC1), where they can be used to investigate inhibition of histone deacetylases.
A second fluorophore used in enzyme assays is 4-methylumbelliferone. An example is found in the application note “CLARIOstar determines activity of a moss-produced human acid alpha-glucosidase (GAA) in a fluorescence-based assay”. The CLARIOstar Plus multi-mode microplate reader makes fluorescent enzyme activity analysis easy. The Enhanced Dynamic Range technology allows reading over a dynamic range of 8 decades. This way, the increasing signal in enzyme activity assays is easily recorded without the risk of choosing a wrong gain or time-consuming instrument optimization. The instrument comes with a free-of-charge analysis software with integrated enzyme kinetic analysis covering Michaelis-Menten and Lineweaver-Burk modelling (Enzyme kinetic measurements performed on a BMG LABTECH microplate reader).
The fluorescence of naturally occurring amino acids like tryptophan can also be used to measure conformational changes and enzyme activity of proteins. Some examples of applications are described in our blog post Tryptophan fluorescence: nature’s probe. Tryptophan fluorescence is useful for example to screen for new therapeutics for drug targets like G-protein-coupled receptors, one of the most popular targets for approved drugs on the market today.
Proteins aggregate if they are misfolded and aggregation correlates with diseases such as Alzheimer’s, Parkinson’s, and prion disease. The analysis of the formation of molecular aggregates helps diagnose and understand these diseases, as well as test potential drugs. The two fluorophores bis-ANS and Thioflavin T (ThT) increase their fluorescence intensity upon interaction with protein aggregates. Bis-ANS preferably interacts with amorphous aggregates as described in the application note “Use of a BMG LABTECH microplate reader to monitor amyloid formation”.
ThT binds to fibrils and is mainly used in kinetic assays. If the sample contains aggregation-inducing molecules, the recombinant proteins aggregate, and fluorescence increases (fig. 10).
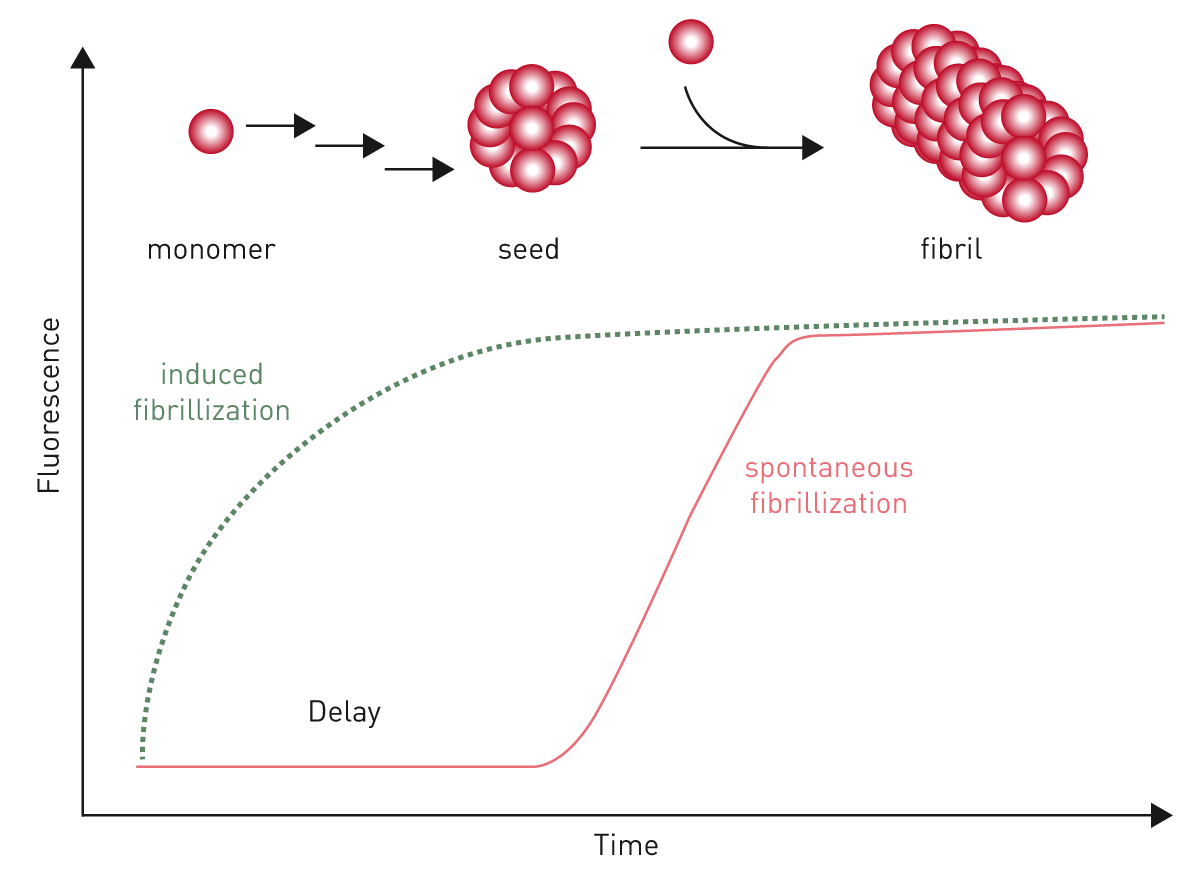
The application note “Real-time quaking-induced conversion assay for prion seeding” explains how the assay is used for detecting the prion diseases scrapie in hamster brain homogenates. As these aggregation assays require long-term shaking, BMG LABTECH's microplate readers can be equipped with very robust and durable transport systems.

Most flexible Plate Reader for Assay Development
Powerful and most sensitive HTS plate reader
Flexible microplate reader with simplified workflows
Upgradeable single and multi-mode microplate reader series


