Introduction
The measurement of protein stability is essential for elucidating protein function in vivo. For example, therapeutic proteins require optimal formulation to improve shelf life and a high-throughput stability measurement would enable many combinations of excipients to be rapidly tested for their effect on protein stability.
Measurements of protein stability are often obtained indirectly by monitoring protein aggregation or residual activity after incubation at elevated temperatures. Both screens rely on irreversible inactivation of the protein upon unfolding. Although these indirect screens have been applied successfully, they may not easily distinguish differences in stability for proteins that spontaneously refold when returned to the activity-assay conditions.
The unfolding transition of proteins can be observed by measuring their tryptophan fluorescence upon perturbation with a chemical denaturant or a temperature shift. Here we describe an unfolding procedure using BMG LABTECH’s microplate reader with titrating syringe pump. Protein unfolding transitions were monitored by tryptophan fluorescence at 340 nm and assessed using bovine and equine cytochrome c (cyt c), as well as bovine serum albumin (BSA) stabilised with various amounts of palmitic acid. Unfolding curves generated by the serial addition of denaturant into single wells, allowed high-throughput stability screens capable of identifying protein variants with unfolding midpoint differences of 0.15 M denaturant concentration or larger. Such a method would be suitable for screening large numbers of proteins or formulation conditions to rank the order of protein stability.
Materials & Methods
Stability measurement
All protein unfolding transitions were measured in F-type, polystyrene, 96-well microplates (Greiner BioOne) using a fluorescence BMG LABTECH microplate reader with one injector. Protein unfolding was monitored by intrinsic tryptophan fluorescence using a 340 - 10 nm emission filter with excitation at 280 nm.
Protein-unfolding curves were obtained by the serial addition of buffered denaturant stock to 50 μL protein solution per well, containing 20 μg cyt c or 40 μg BSA. Each addition was followed by 30 seconds of mixing by orbital shaking at 350 rpm, and 15 minutes of equilibration prior to taking measurements.
Results & Discussion
The incubation times for each addition of denaturant were optimised to ensure that cyt c and BSA unfolding both reached equilibrium. It was determined empirically that 15 minutes between injections were sufficient for both cyt c and BSA to fully equilibrate, resulting in serial addition experiments that took up to 10 hours for 25 denaturant concentrations. Conformational transition curves for cyt c from bovine and equine heart, are plotted as nine replicates performed in parallel (Fig. 1).
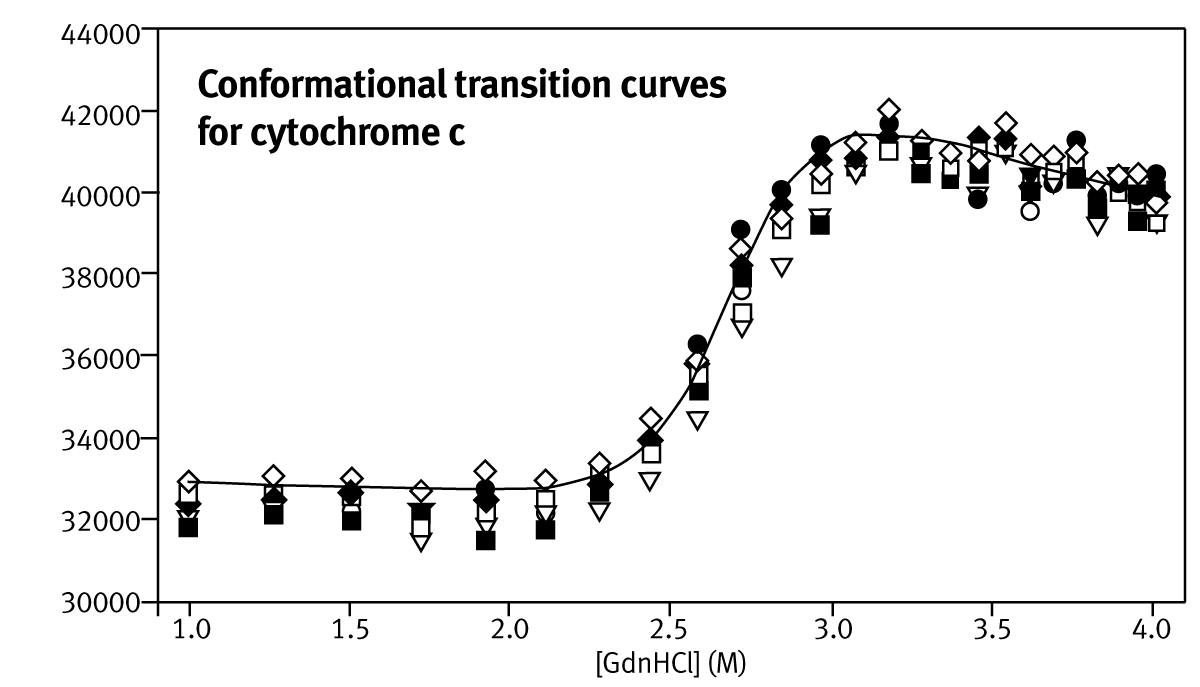
Transition mid-points, C1/2, and mG-values (estimated with non-linear fitting) are summarised in Table 1. The reported stability values are averages of estimates for each of the nine parallel stability analyses.
The inter-run variability was calculated from three separate experiments conducted on separate days, using different stock and protein solutions (Table 1). Mid-points of unfolding for proteins can be estimated to an accuracy of ±0.15 M, but values of mG are significantly overestimated.
Table 1: Summary of stability data for oxidized cytochrome c (cyt c)
| C 1/2 (M) |
mG (kcal/mol) |
ΔG (kcal/mol) | %CV | |||
|
C1/2 |
mG
|
ΔG
|
||||
| Bovine | 2.50 (0.08)a | 6.19 (1.83)a | 14.26 (4.60)a |
2.25b |
22.66b
|
49.09b
|
| Equine | 2.67 (0.14)a | 4.72 (1.19)a | 11.83 (1.35)a |
4.73b |
4.01b
|
6.56b
|
a Standard deviations of the population calculated from nine datasets.
b Percentage coefficient of variation calculated from twenty-seven datasets.
The ability to rank protein stabilities in terms of C1/2 is sufficient for directed evolution screens, where the key requirement is to find enzymes that resist unfolding under the conditions required in a bioreactor. It is also useful for therapeutic formulation in which a combination of excipients is desired that improve the resistance of the protein to unfolding. The large error on mG-value calculation (6.2 ±1.8 kcal·mol-1·M-1 for bovine cyt c) is a result of the inaccurate determination of the pre-and post-unfolding baselines. Unfolding curves of various BSA:palmitate preparations using GdnHCl as denaturant are shown in Figure 2.
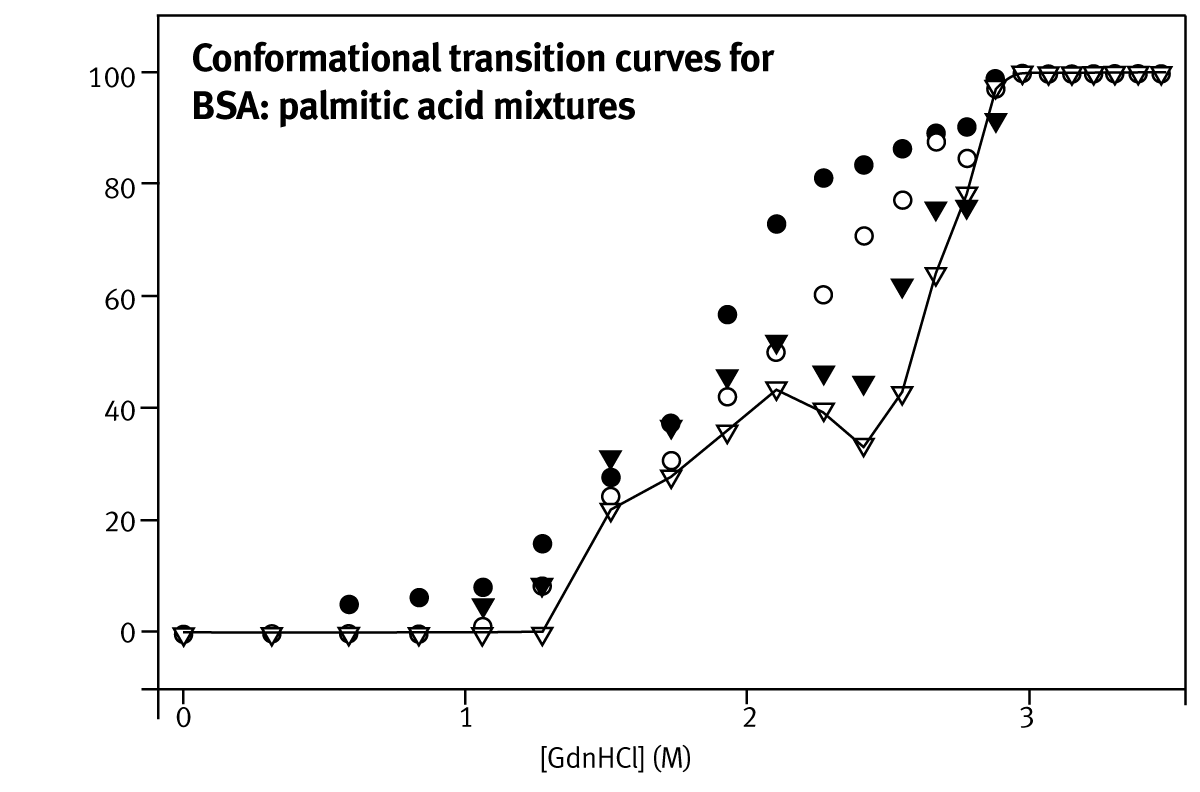
BSA unfolding curves at high-palmitate concentrations display at least one intermediate in the unfolding pathway, as seen from the deviation from a simple two-state unfolding curve. This intermediate is attributed to the early unfolding of the less stable C-terminal part of the protein. Fatty acid molecules partially stabilise the intermediate, increasing the mid-point of urea induced unfolding.
Three simple numerical methods were used to quantify the stability of the different BSA preparations: i) the unfolding datasets were fitted to a two-state transition model, calculating an approximate estimate for a global C1/2-value; ii) the areas below the unfolding curves were calculated using the trapezoidal rule; iii) the denaturant concentration at 60% or 70% unfolding was calculated using a linear equation defined by the two data points either side of the arbitrary set value. Figure 3 depicts the stability estimates using these techniques.
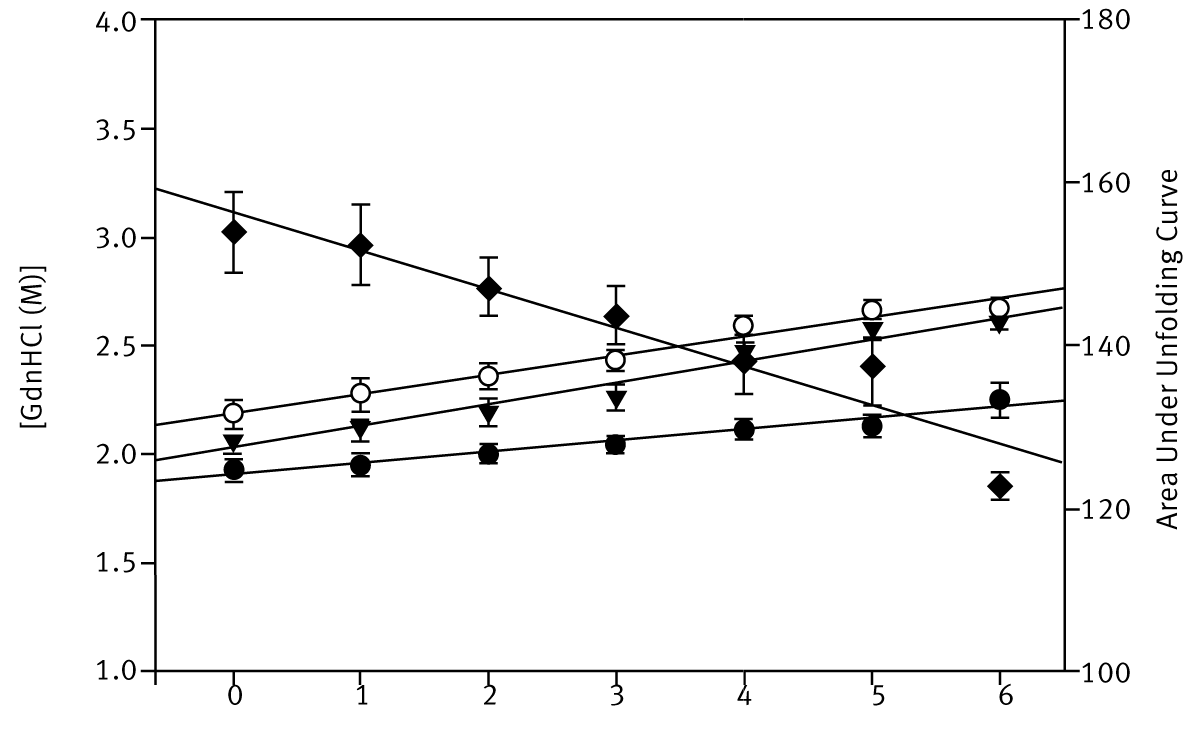 All three numerical methods can confidently determine the order of BSA stabilities present at four palmitate concentrations. Interestingly, the fluorescence unfolding curves for BSA in GdnHCl in microplates (Figure 2) can also detect the presence of intermediate states that occur as three palmitate molecules bind to each BSA molecule, i.e. at high palmitate concentrations.
All three numerical methods can confidently determine the order of BSA stabilities present at four palmitate concentrations. Interestingly, the fluorescence unfolding curves for BSA in GdnHCl in microplates (Figure 2) can also detect the presence of intermediate states that occur as three palmitate molecules bind to each BSA molecule, i.e. at high palmitate concentrations.
Conclusion
We have developed and characterised an affordable high-throughput screening method for the direct measurement of the stability of proteins in a commercially available microplate reader. The method generates unfolding curves in microwells by autotitration of denaturant and measuring the resulting changes in tryptophan fluorescence. The method is useful when screening for changes in C1/2 values of greater than 0.15 M. Combined with a suitable numerical data analysis technique it can be a powerful tool for screening mutant biocatalyst libraries for improved stability.