
PHERAstar FSX
Powerful and most sensitive HTS plate reader


Time-resolved fluorescence (TRF) is a spectroscopic technique that measures the time-dependent decay of fluorescence intensity following excitation.
Fluorescence detection can be mostly divided into two types of measurements: steady-state fluorescence and time-resolved fluorescence. The main difference between these two methods is based on the nature and properties of the fluorescent molecules (fluorophores) used and the consequent time of detection.
Steady-state fluorescence is the most widespread fluorescent detection mode and is commonly referred to as “fluorescence intensity”. Standard fluorophores (e.g. fluorescein, rhodamine, etc.) emit light at a specific wavelength within nanoseconds upon excitation. This very short time gap between excitation and emission allows the almost concomitant detection of the fluorescent emission signal with the excitation of the sample.
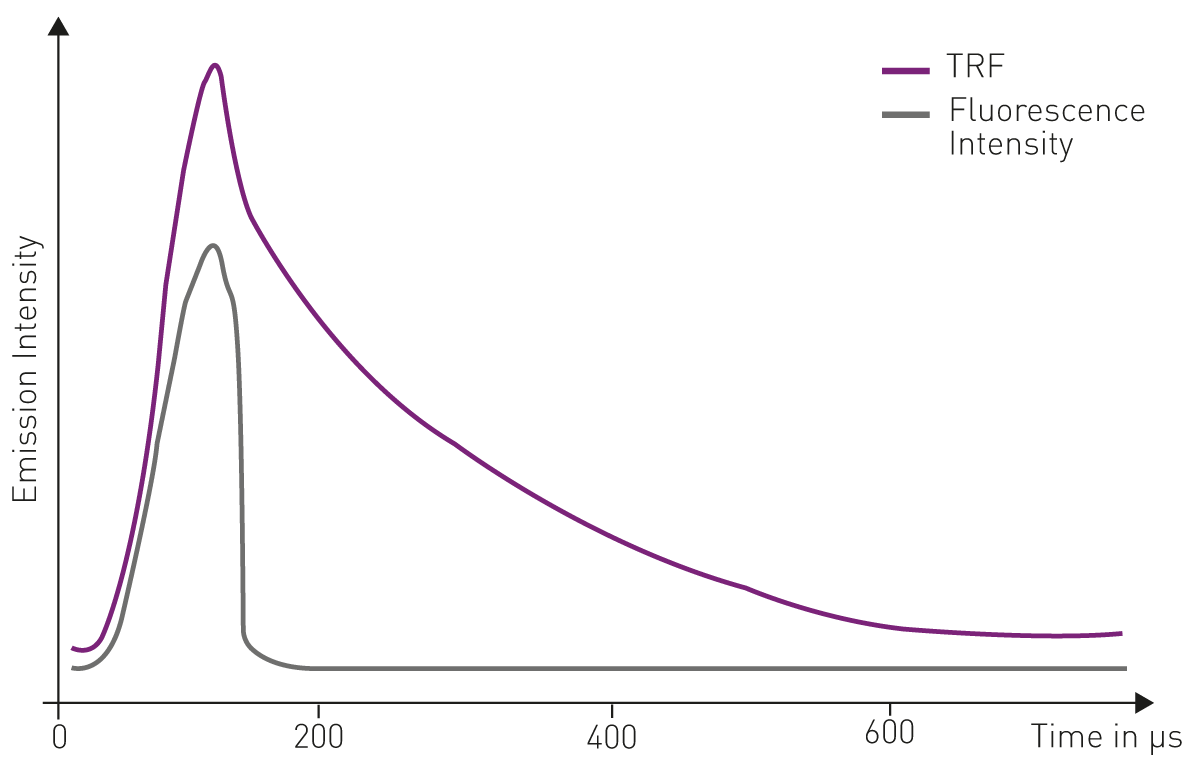
The second type, time-resolved fluorescence (TRF), is monitored as a function of time upon excitation. In contrast to steady-state fluorescence intensity, time-resolved fluorescence is based on the detection of intensity decays and/or on the delayed detection of the emission signal upon excitation. In time-resolved fluorescence measurements, the excitation light pulse is shorter than the decay time of the fluorescent signal. Time-resolved fluorescence detection can only be achieved when the emission signal of the fluorophore is prolonged to the micro- or even milli-second range and not short-lived within nanoseconds as for common labels (fig.1).
Lanthanides (Ln) are a group of uniquely fluorescent metallic chemical elements often collectively known as “rare earth elements”. Lanthanides have very low absorption (excitation) coefficients and slow emission rates. This results in prolonged fluorescence decay times between 0.5 to 3 milliseconds (long lifetimes). All lanthanide elements form trivalent cations (Ln3+) and display emission in an aqueous solution. Additionally, their emission peaks are very sharp and narrow with a large Stokes shift.[1]
Lanthanides possess favorable properties as biochemical probes. Originally, they have been employed in biological systems as luminescent probes for calcium. In fact, lanthanide luminescence has been demonstrated to be a sensitive sensor for Ca2+ binding sites in proteins.[2]
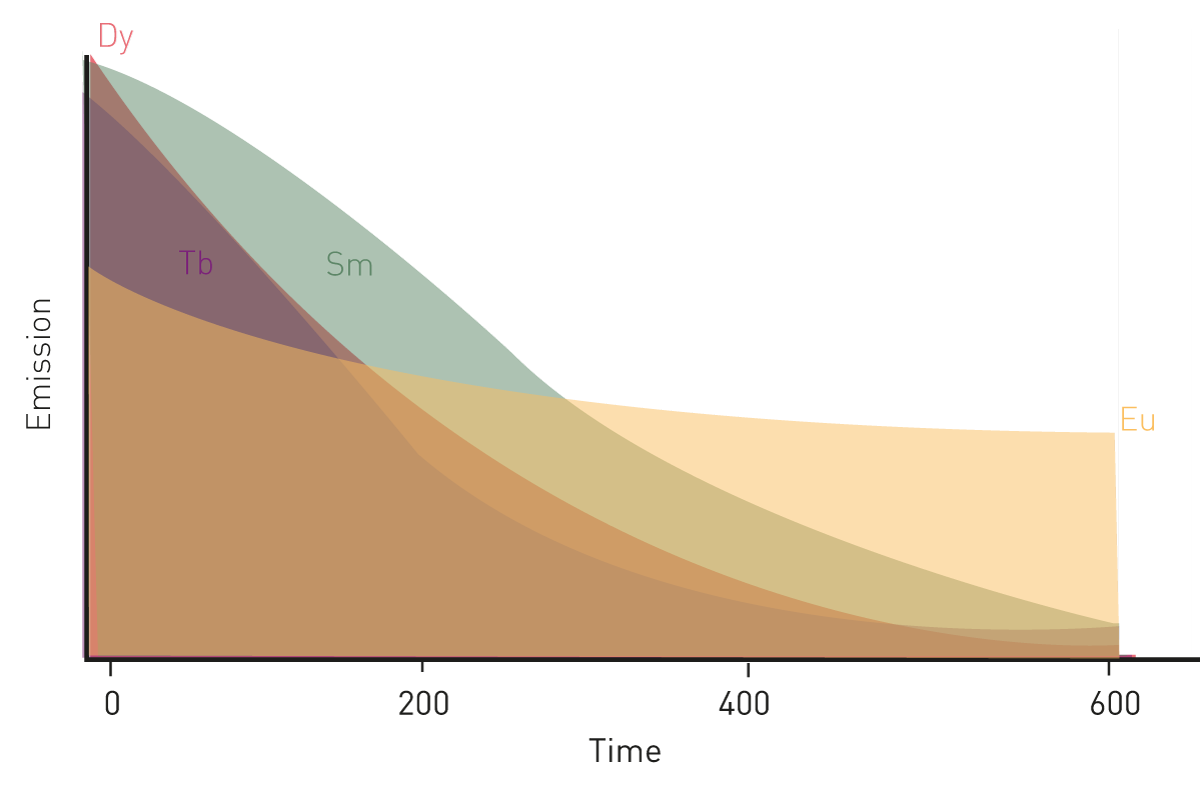
Their prolonged fluorescence decay makes them ideal fluorophores for time-resolved fluorescence applications. Four of them, europium, terbium, samarium, and dysprosium have found widespread use in the life sciences, in particular in time-resolved fluorescence immunoassays, with europium and terbium being the most commonly used (fig.2).
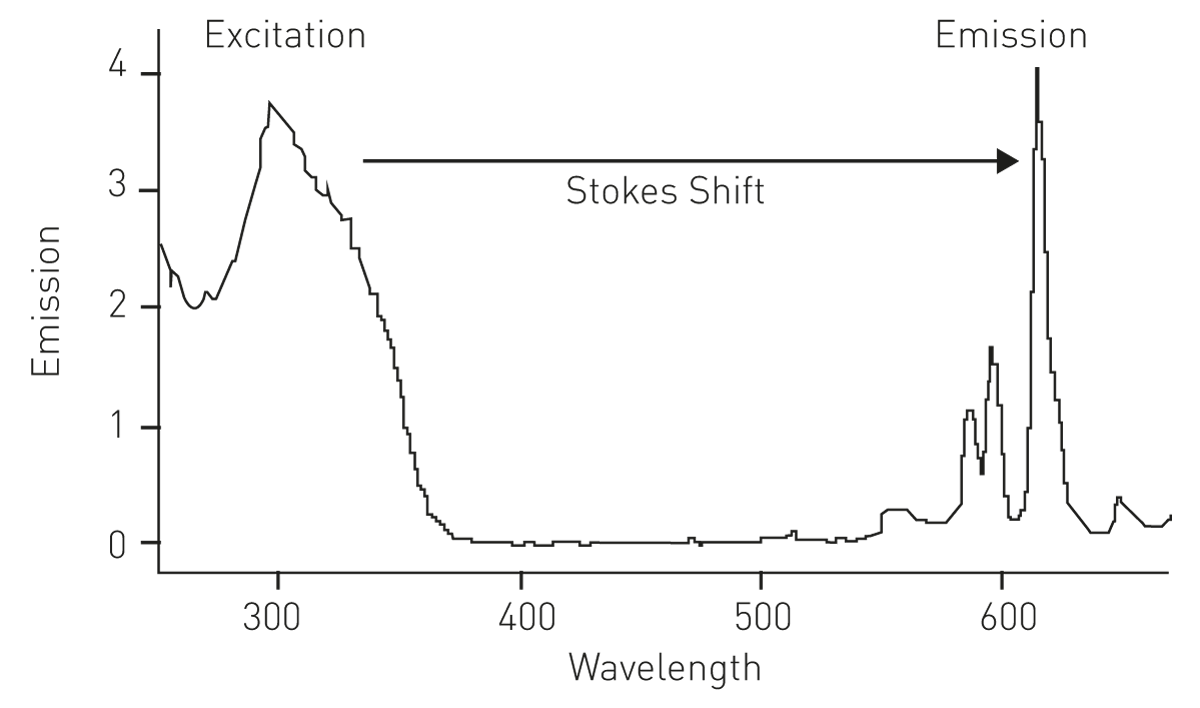
Europium ions (Eu3+) in particular, are frequently used as labels for time-resolved fluorescence detection in immunological assays. Besides the long emitting fluorescence, europium displays a large Stokes shift (290 nm) with no overlap between the excitation and emission spectrum, and with only 10 nm bandwidth, a very sharp emission spectrum at 615 nm (fig.3).[3]
As the emission of lanthanides is usually too weak for time-resolved fluorescence applications, they are generally not directly excited, but are usually embedded in a sort of light-collecting “cage”. This cage, most commonly a chelate or a cryptate, allows both energy collection and energy transfer to the lanthanide ions, resulting in higher emission intensities (fig.4). It is worth to be mentioned that the excitation spectrum of the cage-lanthanide complex reflects the absorption spectrum of the “cage” and not of the lanthanide itself.[4]
In addition to a higher emission signal, chelation makes lanthanide ion conjugation to biological components (e.g. antibodies, receptors, ligands, etc.) possible, a mandatory requirement for several time-resolved fluorescence applications.
![Fig. 4: example of a europium-chelate complex in fluorescent state. Modified from [5].](https://www.bmglabtech.com/hubfs/1_Webseite/5_Resources/ABC/Detection%20Modes/time-resolved-fluorescence-fig4.webp)
Time-resolved fluorescence is detected by applying a delay after pulsed excitation and measuring the emission signal only during a defined time window, capturing long-lived emission while excluding short-lived background signals.
As the lanthanide-chelate/cryptate complex is typically excited at 337 nm, a xenon flash lamp or a specific laser is used as a light source for time-resolved fluorescence detection. Multi-mode plate readers are equipped with a broadband xenon flash lamp as it offers more flexibility for multiple detection methods. High-end readers may offer in addition an optional dedicated time-resolved fluorescence excitation laser. The laser focuses more energy on the specific excitation wavelengths of lanthanides and possibly leads to better results with better discrimination between low and high signals. However, given the sharp excitation at around 337 nm, this is just a single-purpose light source that cannot be used as an excitation source for other detection methods but time-resolved fluorescence.
Regarding wavelength selection, both filter- and monochromator-based plate readers can be employed for the detection of time-resolved fluorescence assays. As filter-based readers are usually more sensitive than monochromators because of higher light transmission, they are recommended for TRF assays with a limited photon yield. Typically, time-resolved fluorescence energy transfer assays are particularly challenging for monochromator-based readers.
Photomultiplier tubes (PMTs) are used as detectors. Because of the long-emitting properties of lanthanides, in time-resolved fluorescence, the PMT detector is turned on after the excitation has occurred. This allows the short-lived autofluorescent signal to fade out. The intensity of the emission signal is then integrated as a function of time for a specific time window. These two parameters are referred to as “integration start” and “integration time” and are usually in the microsecond range (fig. 5).
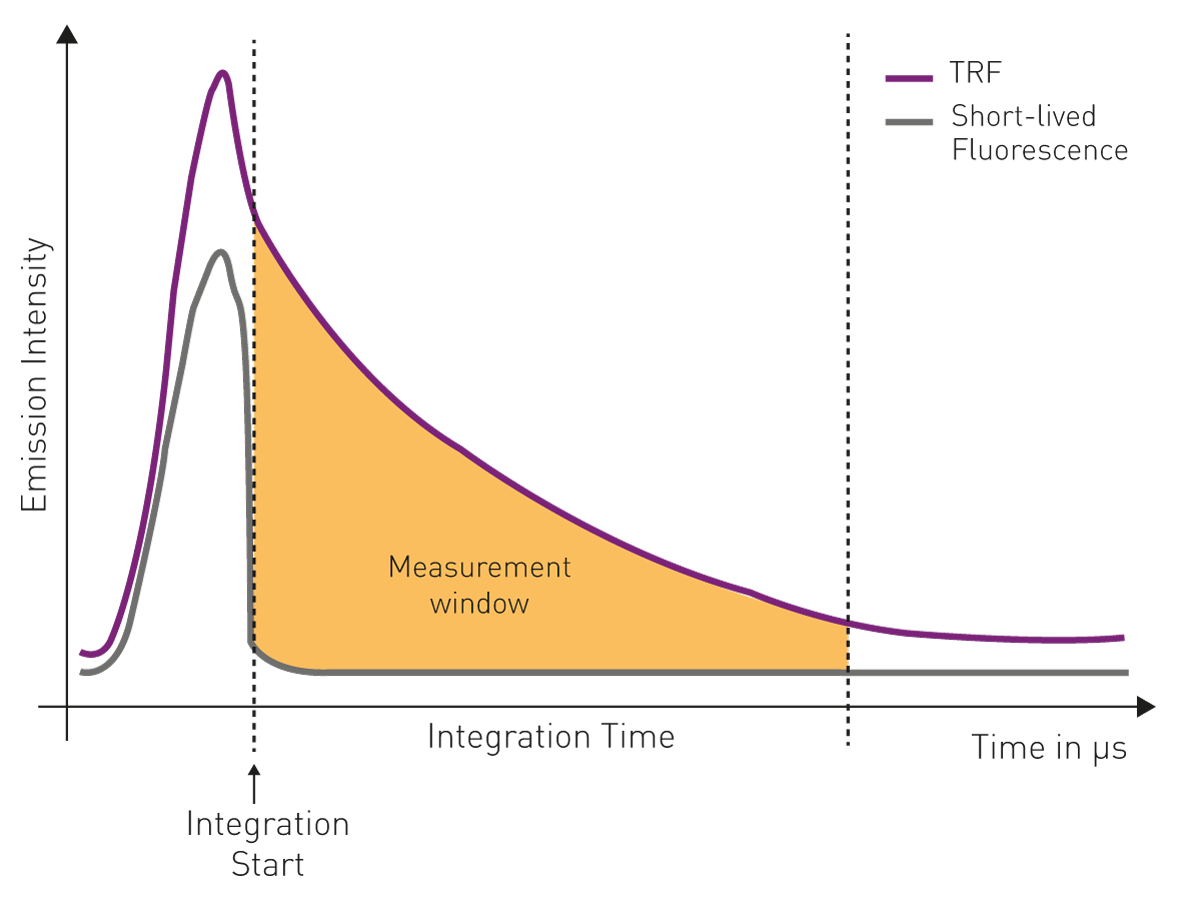
High-end microplate readers can also be equipped with so-called photon-counting PMTs for time-resolved fluorescence. While normally microplate readers simply give an integration value for the area under the curve during the integration time, time-resolved fluorescence -dedicated photon-counting detection monitors the whole decay curve of the lanthanide.
On the PHERAstar FSX reader, photon-counting detection allows the measurement and display of the emission decay curve with a time resolution of 2 microseconds. This unique feature called Decay Curve Monitoring simplifies time-resolved fluorescence assay development, helping to optimize timing parameters and thus improving signal detection and reducing background noise.
Time-resolved fluorescence immunoassays are used to quantify specific molecules such as proteins or cytokines by relying on target recognition and binding by specific antibodies that have been labelled with a fluorophore. Analysis and quantification of the fluorescent signal indirectly provide information on the target molecules. Immunoassays are quantitative, highly sensitive, and provide the possibility for multi-parameter detection (multiplexing).
Similar to the classical ELISA assays, time-resolved fluorescence immunoassays commonly use a capture antibody bound to the bottom of the microplate well. As the samples are incubated in these wells, the antibody captures the target molecule on the plate. After removal of an unbound sample by washing the plate, a second antibody covalently bound to a lanthanide chelate, most commonly europium, is added. This binds the target molecule while the non-bound second antibody is washed away. The amount of lanthanide labelled antibody is proportional to the concentration of the target molecule in the sample. Time-resolved fluorescence immunoassays can be performed as direct or competitive assays.
A dissociative enhancement step is required to free the europium molecules from their “cage” on the antibody because of three reasons: first, lanthanides have poor light absorption; second, they are rarely directly excited; and third, lanthanide chelates are only poorly fluorescent when conjugated to biological components (antibodies in this case).
The dissociation is promoted by the addition of a specific solution commonly referred to as an “enhancement solution”. Besides dissociating the lanthanide, the solution promotes the formation of a new chelate “cage” that incorporates the chromophore necessary for the excitation of the lanthanide. The chelator-chromophore in the enhancer solution is used to absorb and transfer the excitation light to the lanthanide, significantly increasing the intensity of the emission signal (fig.6).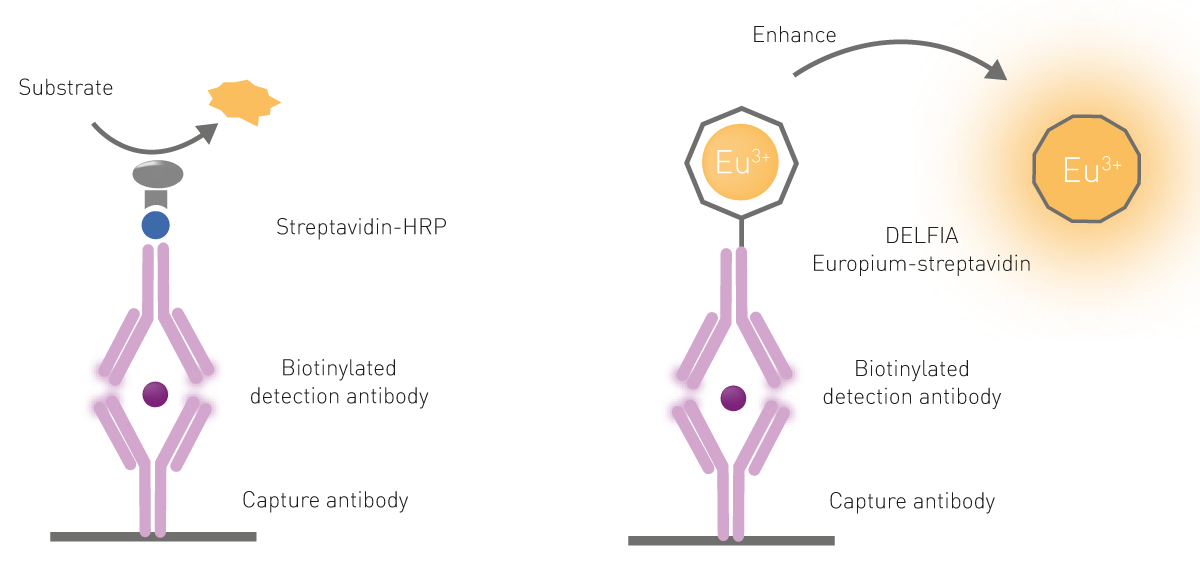
Samples are excited by a pulse of light at a specific wavelength, usually 337 nm. The detection is time-gated on the decay of the autofluorescence signal. This means that time-resolved fluorescence detection starts only after exhaustion of the short-lived autofluorescence signal (microseconds). The detected emission signal is integrated for a specific time window and data are measured as integrated intensity, not time decay. As the amount of analyte is proportional to the time-resolved emission signal, it can be easily quantitated with the use of a standard curve.
One of the most commonly used time-resolved fluorescence immunoassays is DELFIA®.
DELFIA (Dissociation-Enhanced Lanthanide Fluorescent Immunoassay) is a heterogeneous time-resolved fluorescence wash-based assay developed on a similar principle and workflow as ELISAs. It is said to overcome the typical limitations of ELISA assays, providing a wider dynamic range and stable signals that can be measured up to months after the execution of the assay. For DELFIA time-resolved fluorescence assays, plate readers have to be equipped with TRF detection with excitation at 337 nm and emission at 615 nm.
In DELFIA assays, capture antibodies are bound to the microplate. Upon sample addition and a series of wash steps to eliminate the unbound sample, a europium-labelled detection antibody is added. Finally, an enhancement solution is added after a final series of washes. As mentioned above, DELFIA time-resolved fluorescence assays require a dissociative enhancement step to produce a fluorescent signal. This dissociation promotes the formation of a new highly fluorescent chelate in a stable micellar solution. An example of a DELFIA immunoassay is shown in the application note “Time-Resolved Fluorescence (TRF) immunoassay in 384-well format using a matched antibody pair kit and the PHERAstar FSX”.
Although robust and very sensitive, DELFIA assays are not suited for high-throughput screening, as the procedure involves binding, incubation, and washing steps.
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Flexible microplate reader with simplified workflows
Upgradeable single and multi-mode microplate reader series


