Introduction
Fluorescence polarization (FP) provides a useful tool to screen for inhibitors of protein-peptide interaction.
FP gives a measure of the proportion of peptide found in the bound state in a homogeneous format. However compound interference and non-specific gross structural changes to the protein can give rise to a large number of false positives, which are only identified in later stage biochemical assays. Strategies to eliminate these at an early stage of screening will accelerate the hit to lead process.
Here we present data from an FP screen configured on a BMG LABTECH microplate reader for the interaction of the retinoblastoma tumor suppressor protein (pRB) with E2F peptide. Fluorescein-tagged E2F peptide was used to screen 10,000 small drug like molecules. Hit confirmation strategies based on fluorescence interference and specificity were developed.
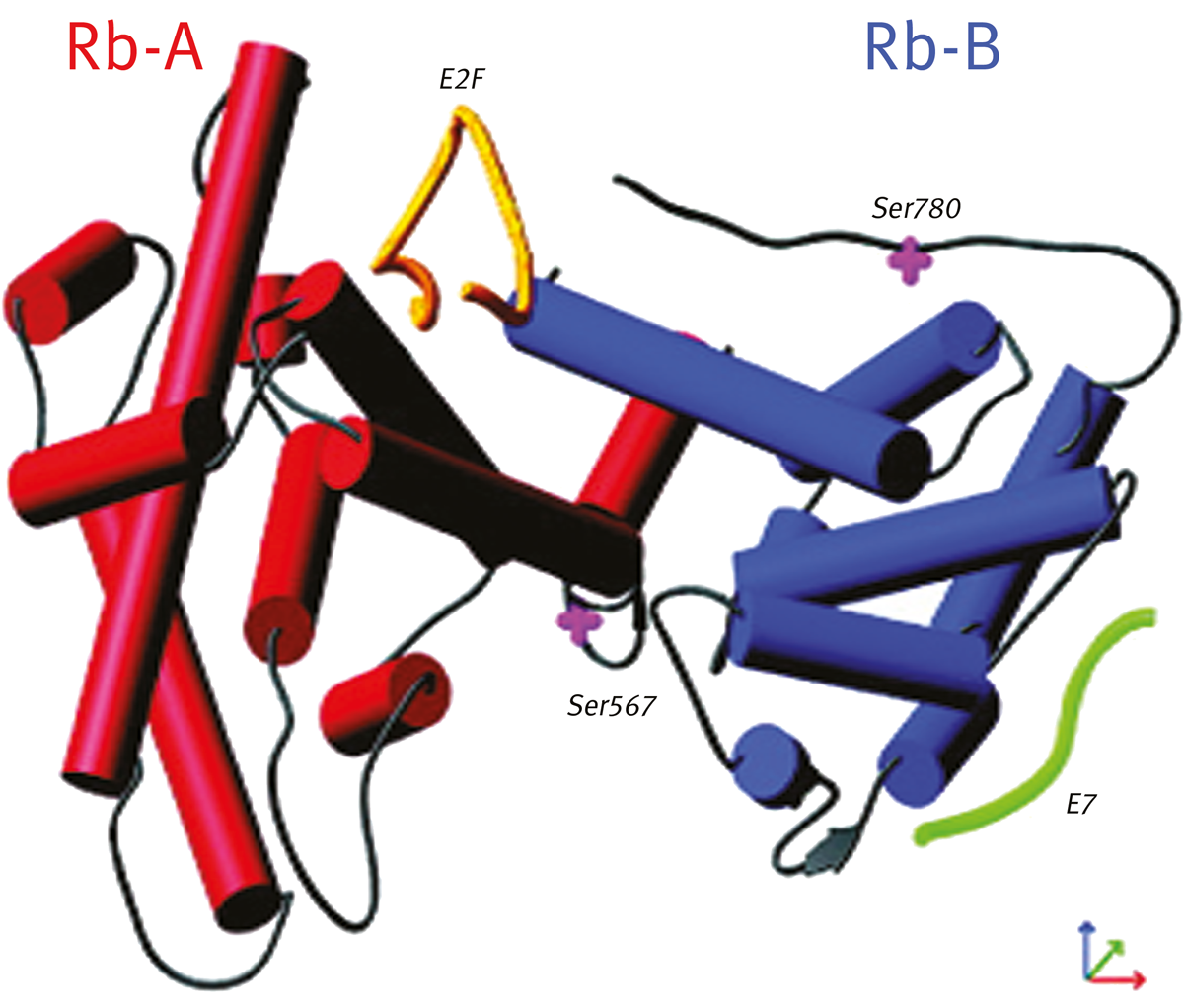
An FP screen was configured for the interaction of recombinant pRb A/B domains with E2F peptide (depicted in yellow in figure 1). In addition, a second peptide binding site (E7, depicted in green), was utilised as an internal control for non-specific inhibitors. Fluorescein-E7 and rhodamine-E2F labelled peptides were used for hit confirmation.
Materials & Methods
Peptides were synthesised and fluoro-tagged. Before use peptides were dissolved in assay buffer (50 mM Tris HCl, pH 7.0, 100 mM NaCl, 10 mM DTT, 0.05% NP-40). Typical titration binding curves of pRb with the fluorolabelled peptides are shown (mean±sem, n=3) in figure 2. Fluorescein-E2F showed the greatest degree of polarization, and consequently the best signal to noise. It was chosen as the label of choice for a primary screen. Data were fitted to a one site binding model using Graphpad prism.
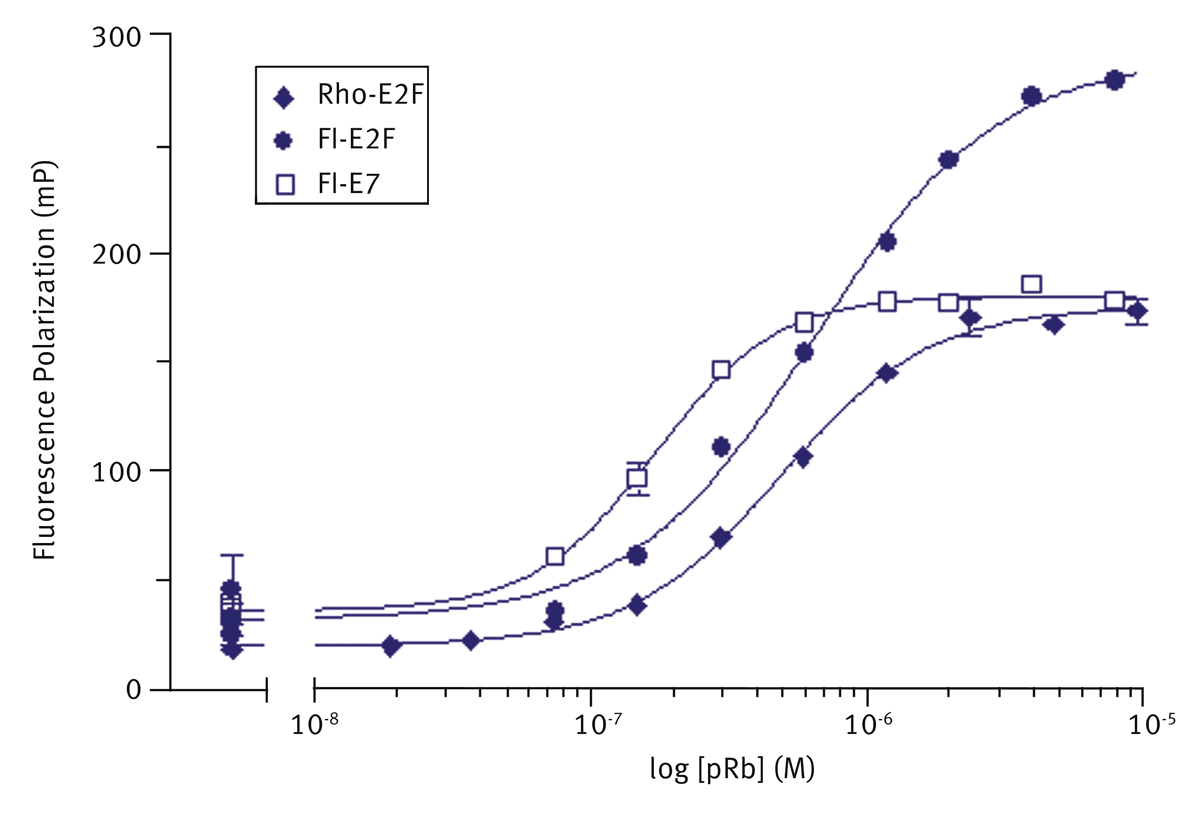
Kd values of (450±70) nM and (380±50) nM were calculated for fluorescein and rhodamine labelled E2F, which were similar to Kd determined for unlabelled peptide using isothermal calorimetry. Fluorescein-E7 showed tightest binding with Kd = (130±20) nM.
The assay was optimised in 384-well black plates (Matrix) and automated using a Beckman Fx liquid handling robot. 1 μM pRb in 50 mM Tris HCl, pH 7.0, 100 mM NaCl, 10 mM DTT, 0.05% NP-40 was mixed with 40 μM compound (4% DMSO) and 0.4 μM fluorescein-E2F (final concentrations). Total reaction volume 50 μL. Controls from a test screen of 10,000 compounds are shown in figure 3.
BMG LABTECH microplate reader settings
- Fluorescein detection: ex 485-12, em 520-30 nm
- Rhodamine detection: ex 545-7, em 580-12 nm
- Default general settings: 50 flashes, 1.0 s positioning delay, 1 cycle
Results & Discussion
Polarized and depolarized signal from fluorescein- E2F with and without pRb present are shown in figure 3 (solid and open blue circles respectively). Specific disruption of binding by E2F protein and peptide are also shown.
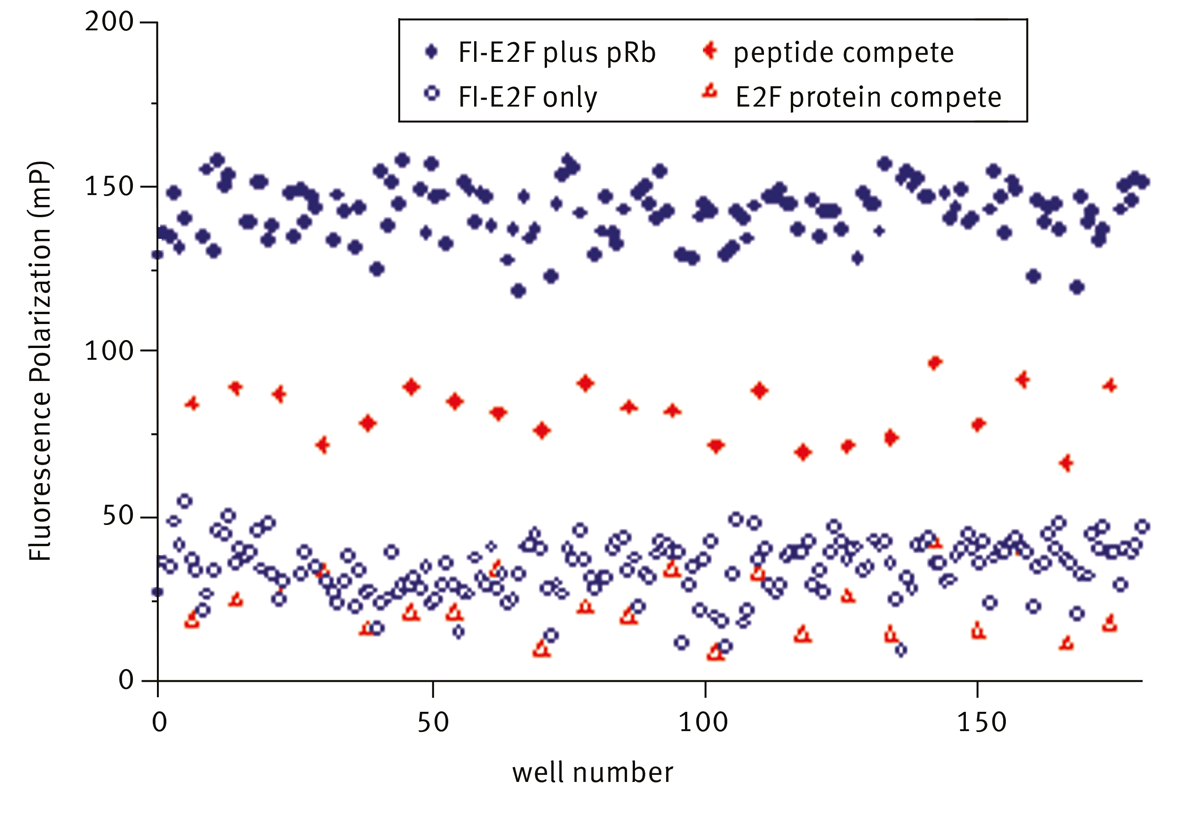
Addition of E2F protein completely displaces Fl-E2F (open red triangle) and the signal is reduced to that of free fluoro-peptide alone. Addition of unlabelled-E2F peptide (solid red diamond) at a concentration which gave 50% inhibition is clearly separated from the control populations. Hits were identified as compounds which reduced the polarization signal to less than mean-3sd of the fluorescein-E2F: pRb control. A summary of the screen data is shown below in table 1.
Table 1: Summary of screen data.
|
Assay Parameters
|
Signal: Noise
Signal: Background
Z'
|
6.9
4.8
0.67
|
|
Test Screen 10,000
|
Z'
Hit Rate
|
0.45
0.93 %
|
A large proportion (37.5%) of the hits selected from the primary screen were coloured compounds which significantly altered the fluorescence intensity signal, and were potentially interfering with the assay. All hits were included in the hit confirmation assays.
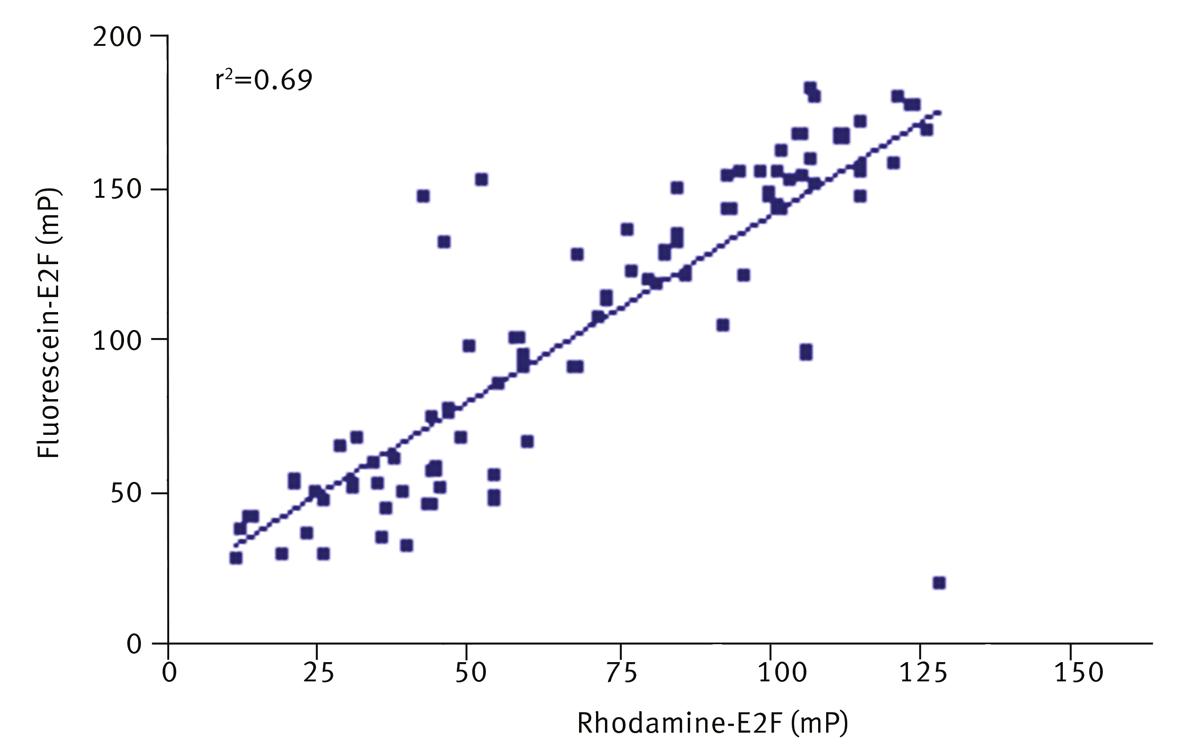 Hits were replated from master stocks and retested against fluorescein-E2F and rhodamine-E2F. A correlation (r2=0.69) between inhibition of fluorescein-E2F and rhodamine-E2F was observed (figure 4) with a hit confirmation rate of 78%. Notably, 60% of compounds which were potentially interfering with the fluorescein signal were inhibitors with rhodamine-E2F assay, without affecting rhodamine fluorescence intensity signal. Suggesting that deselection of compounds on the basis of fluorescence interference can lead to loss of real inhibitors.
Hits were replated from master stocks and retested against fluorescein-E2F and rhodamine-E2F. A correlation (r2=0.69) between inhibition of fluorescein-E2F and rhodamine-E2F was observed (figure 4) with a hit confirmation rate of 78%. Notably, 60% of compounds which were potentially interfering with the fluorescein signal were inhibitors with rhodamine-E2F assay, without affecting rhodamine fluorescence intensity signal. Suggesting that deselection of compounds on the basis of fluorescence interference can lead to loss of real inhibitors.
Finally, the hits were tested against a second peptide binding site, fluorescein-E7 peptide at 400 nM, as shown in figure 5. The results were compared to inhibition of E2F and a scatter plot is shown.
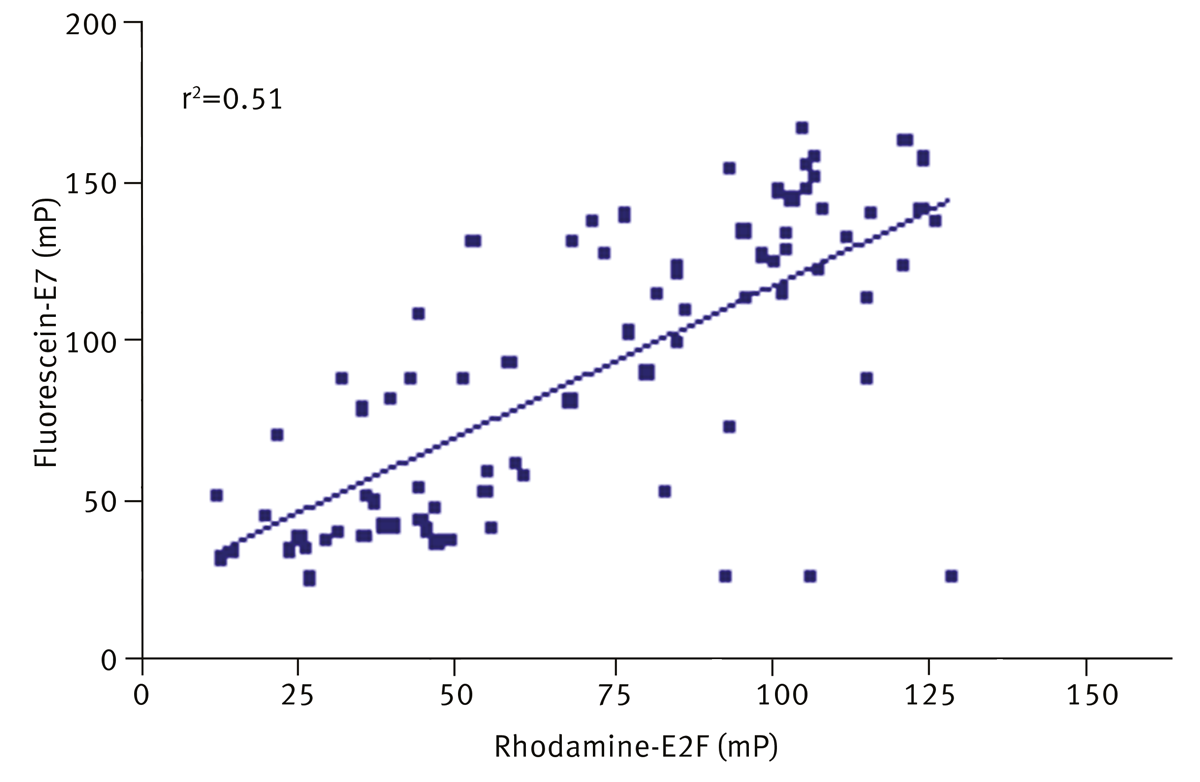 A weak correlation was observed (r2=0.51), with 72% of the inhibitors of E2F also inhibiting fluorescein-E7. These compounds were excluded as non-specific inhibitors and were not taken forward in subsequent biochemical assays. 14 hits were taken forward from a total of 80 hits identified in the primary screen.
A weak correlation was observed (r2=0.51), with 72% of the inhibitors of E2F also inhibiting fluorescein-E7. These compounds were excluded as non-specific inhibitors and were not taken forward in subsequent biochemical assays. 14 hits were taken forward from a total of 80 hits identified in the primary screen.
Conclusion
Binding of fluorescein-E2F to pRb showed a greater degree of polarization compared to binding of rhodamine-E2F, and consequently, a better signal to noise ratio and Z’ factor could be obtained. It was the label of choice for a primary screen.
Screening of the hits against the second peptide site, E7, identified non-specific inhibitors, which caused gross structural changes to the protein. The combination of rhodamine-E2F and fluorescein-E7 with pRb in the same well allowed rapid selection of specific inhibitors and minimised reagent usage. Identification of these non-specific inhibitors dramatically reduced the down stream work load.