SPECTROstar Nano
Absorbance plate reader with cuvette port


This author profile refers to work created by Dr Andrea Krumm during her tenure at BMG LABTECH - the Microplate Reader Company, where she served as an Applications Specialist contributing scientific content, application notes, and technical expertise. Dr Krumm is no longer employed at BMG LABTECH, but her published materials remain available here for reference and archival purposes. Dr Andrea Krumm is a biotechnology specialist and product manager known for her work in analytical instrumentation and biopharmaceutical analysis. She studied biotechnology and later earned a PhD in cancer biology, focusing on topics such as DNA repair, epigenetics, and tumor biology. After completing her doctorate, she spent several years as an Applications Specialist at BMG LABTECH, where she authored application notes, conducted workshops, and supported scientific customers. In 2020, Dr Krumm joined Tosoh Bioscience GmbH as Product Manager for Analytical Columns.
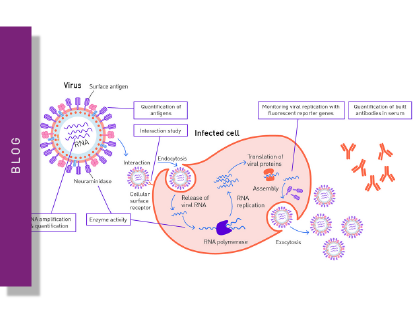
A virus (Latin for poison) is an obligate intracellular parasite of sub-microscopic size with simple structure that can only replicate in living cells of animals, plants and bacteria. Viruses are acellular and consist of nucleic acid surrounded by a protein coat and sometimes a membranous envelope. They divert the machinery and metabolism of host cells to produce and assemble multiple copies of themselves, as they lack metabolic enzymes, ribosomes and are unable to synthesize proteins on their own. Therefore, many virus assays employ host cells to study viral biology.
The replicative life cycle of viruses
Outside infected cells, viruses exist as independent particles called virions. The types of cells that a virus can infect (called host range) are generally limited and specific so is one basis of classification.
Although the viral life cycle differs greatly between species, six basic stages are essential for viral replication:
1. Attachment
The virus attaches through a surface protein to specific receptors present on the host cell membrane. The specificity of the engaged molecule determines the host range.
2. Penetration
Virions enter the host cell through endocytosis or membrane fusion. The infection of bacteria, plant and fungal cells differs from that of animal cells because of the presence of a cell wall.
3. Uncoating
The viral capsid is removed either by degradation or dissociation, releasing the viral nucleic acid inside the host cell.
4. Replication
After release of the genome into the cell, transcription and/or translation is initiated. Specific techniques report on viral replication and its inhibition for possible treatments.
5. Assembly
Virus proteins are post-transcriptionally modified and packaged with newly replicated viral genome into new virions.
6. Release
Viruses are released from the host cell via cell lysis, budding or exocytosis. They can also stay dormant within the infected cell - replication is reduced until reactivation.
Viral structure
Virions consist of nucleic acid and a protein coat called capsid. The capsid is formed by identical protein subunits (capsomeres), which surround and protect the genetic material. In some cases, they have an outer lipid envelope derived from the host cell membrane.
The capsid´s shape helps to morphologically divide viruses. Types include helical or filamentous, icosahedral, enveloped and more complex structures (fig. 1).1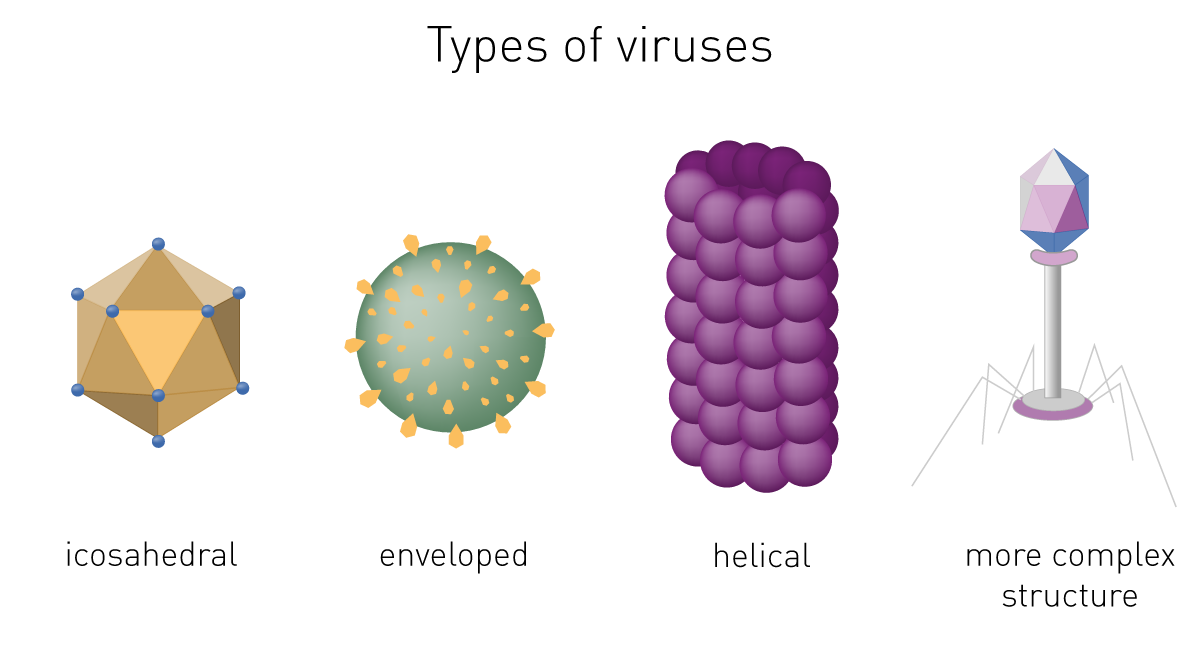
Virus genome – Baltimore classification
The most common classification is based on the viral nucleic acid and its replication. The Baltimore classification distinguishes between DNA and RNA, single- and double-stranded, and whether the genetic information can be used directly or requires transcription or insertion into the host genome (fig. 2).2 As many diagnostic virus detection methods reveal the presence of virus genome, its nature has to be considered when preparing the genomic material for testing.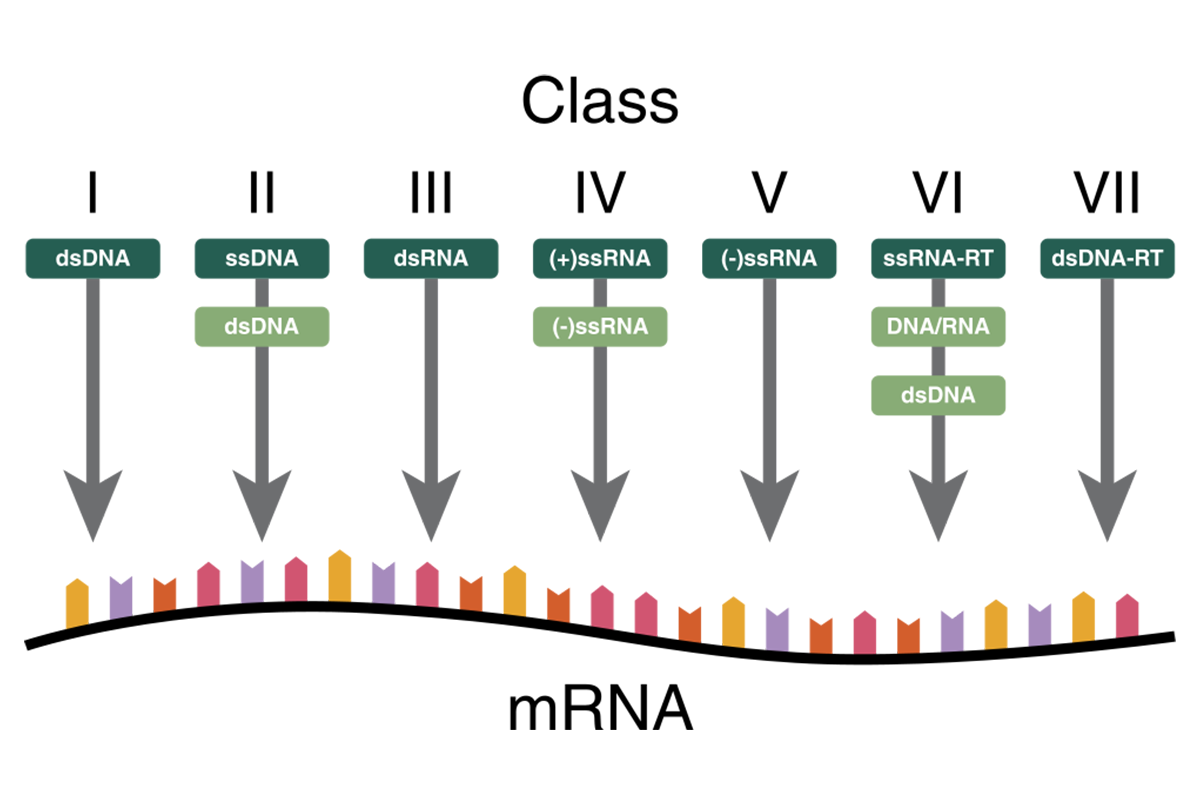
Group I: Double-stranded DNA (dsDNA) viruses
Group I viruses contain dsDNA in linear, circular or circularly permuted (the same genetic information starts and ends at different locations in the genome) form. Most of them replicate in the nucleus using the host translation machinery.
Examples:
Herpes simplex: causes oral and genital herpes - nuclear replication
Adenovirus: causes respiratory infections, cold symptoms - nuclear replication
Smallpox: responsible for the potentially fatal disease smallpox, declared extinct due to vaccination - cytoplasmic replication
Group II: Single stranded DNA (ssDNA) viruses
Group II contains ssDNA and requires a conversion into dsDNA before transcription to mRNA. These viruses mainly infect invertebrates, archaea and fungi. However, one human pathogen is known.
Example:
Parvovirus B19: causes “fifth disease” in children, producing cold-like symptoms and a rash, especially on the cheeks
Group III: Double-stranded RNA (dsRNA) viruses
Viruses in group III have a dsRNA genome. One RNA strand serves as template for the generation of mRNA, using the virus-specific RNA-dependent RNA polymerase (RdRP) and the host translation machinery for translation into proteins.
Example:
Rotavirus: infects small intestine cells, leading to diarrhoea and gastroenteritis in children.
Group IV: Positive sense single-stranded RNA (+ssRNA) viruses
Group IV has a ssRNA with positive polarity as genome. ssRNA is directly translated by the host into proteins that make up the virus replication complex. RNA strands of negative polarity (-ssRNA) are produced and serve as templates for +ssRNA synthesis. The +ssRNA is either translated or encapsulated to leave the host cell.
Examples:
Rhinovirus: major cause for a common cold
Dengue: transmitted by mosquitos, leads to “dengue fever” with fever, headache, vomit, muscle and joint pains
SARS-CoV-2 (Severe Acute Respiratory Syndrome Corona Virus 2): causes the disease COVID-19, first described end 2019 in Wuhan, China. The disease often presents mild symptoms, such as a scratching throat, dry cough and fever but can also escalate to pneumonia, multiple organ failure or even death, the mechanism of which is not fully understood. This virus has spread rapidly and was declared a pandemic in early 2020 by the WHO.
Group V: Negative sense ssRNA (-ssRNA) viruses
Viruses in this group contain ssRNA with negative polarity (complementary to mRNA). RNA-dependent RNA polymerase encoded by the virus can produce mRNA. -ssRNA can be directly converted to mRNA. +ssRNA serves as template to produce the -ssRNA genome.
Examples:
Ebola: causes viral hemorrhagic fever, sore throat, diarrhea, vomit, and has ~50% fatality
Measles: causes childhood disease with high fever, a runny nose, white spots in the mouth and rash - preventable by vaccination
Influenza: causes fever, muscle and joint pain, headache and a runny nose - partly preventable with a yearly updated vaccine
Group VI: Single strand RNA retroviruses (ssRNA-RT)
Group VI viruses contain ssRNA. This is converted to dsDNA by the enzyme reverse transcriptase, transported to the nucleus and inserted into the host cell genome by an integrase. mRNA is produced by transcription of the integrated viral DNA.
Example:
Human Immunodeficiency Virus (HIV): induces the Acquired Immune Deficiency Syndrome (AIDS). No vaccine is currently available, only symptomatic treatments to suppress the virus and reduce transmission.
Group VII: Double-stranded DNA retroviruses (dsDNA-RT)
Group VII retroviruses have dsDNA genomes. They replicate through an RNA-intermediate which is reverse transcribed by virus reverse transcriptase and converted back into dsDNA. This differentiates them from group I dsDNA viruses that employ DNA polymerases for replication.
Example:
Hepatitis B: may be asymptomatic or cause severe liver inflammation. A vaccination for long term immunity is available.
Virus transmission
Viruses spread using living organisms as vectors. Once infected, the organism becomes a reservoir of virions which can be released in bodily fluids. Virions can be exchanged orally, sexually or through contact with contaminated surfaces. After infection, they induce disease by different mechanisms.
The pathogens trigger cell lysis to release virions. If enough cells die, multicellular organisms will start to suffer the effects, depending on the attacked organ or tissue.
In higher organisms, viral infections trigger an immune response meant to eliminate the infection. The first line of defence is the innate immune system which recognises external threats and defends the host in a non-specific manner. A specific response comes from the adaptive immune system, the humoral immunity. This produces specific antibodies that bind and inactivate the intruders, possibly conferring protective immunity to the host. Some pathogens have evolved to evade host immune responses and can hence cause chronic infections.
Treatment of viral infections
Modern medicine combats viral infections mainly through vaccination and drug treatments. Vaccinations confer artificially acquired immunity to a specific virus. This is usually the most effective medical approach as vaccines proactively prevent infection and spread of the virus.
Drug treatments are targeted to reduce viral entry to cells and replication. As viruses rely on cellular metabolic pathways to replicate, drug treatments can cause toxic side effects to the host. Although a more difficult approach, antiviral drugs, through their selectivity and ability to interfere with viral replication, have been shown to be very effective.
Diagnostic methods accurately identify viral infections in patients. This is a prerequisite to control and limit virus propagation through an effective clinical management of the disease1. A precise identification of the ongoing or past infection helps to prevent transmission, to set up an appropriate therapy and to monitor treatment3. Failure in diagnosis can lead to significant human and financial loss.
Virus assays vary in approach, efficacy and cost. Besides performance parameters including accuracy, precision, sensitivity, and specificity which relate directly to the results, simplicity and ease in performing the tests are also important factors. The Turn Around Time (TAT), the interval time between sample registration and result reporting, is also used as a key performance indicator4.
Long-established virus assays for diagnosis
Traditional laboratory identification methods of viral infections focus mainly on optical detection of virions by microscopy techniques or by indirect serologic evidence.
a) Electron microscopy (EM)
EM for a long time has been considered an efficient tool for the direct detection of viruses by visual counting of the viral particles in body fluids, stools or histopathologic samples. Though essential in revealing morphology and appearance, it requires a high virion number in samples and substantial technical skills and expertise by the operator3.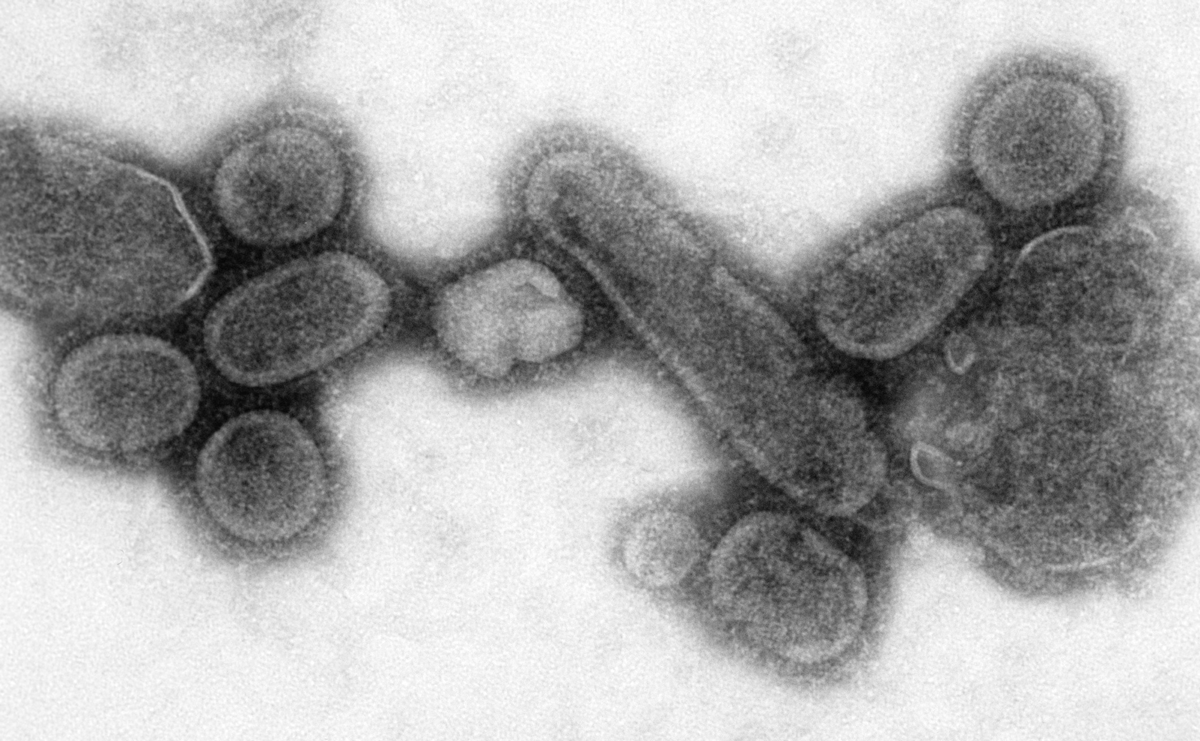
b) Light microscopy
The analysis of viral proliferation within a cell monolayer by light microscopy is a standard diagnostic technique. Changes in cellular appearance like swelling, shrinking and syncytium formation are examined. They indicate the presence of the virus and are defined as cytopathic effect (CPE) 3,5.
Although CPE is usually observed after 5–10 days of incubation, it can vary from 24 hours for herpes simplex to 10–30 days for cytomegalovirus5,6. Incubation time for CPE is an important characteristic of a virus and can be used as a form of characterisation in viral identification.
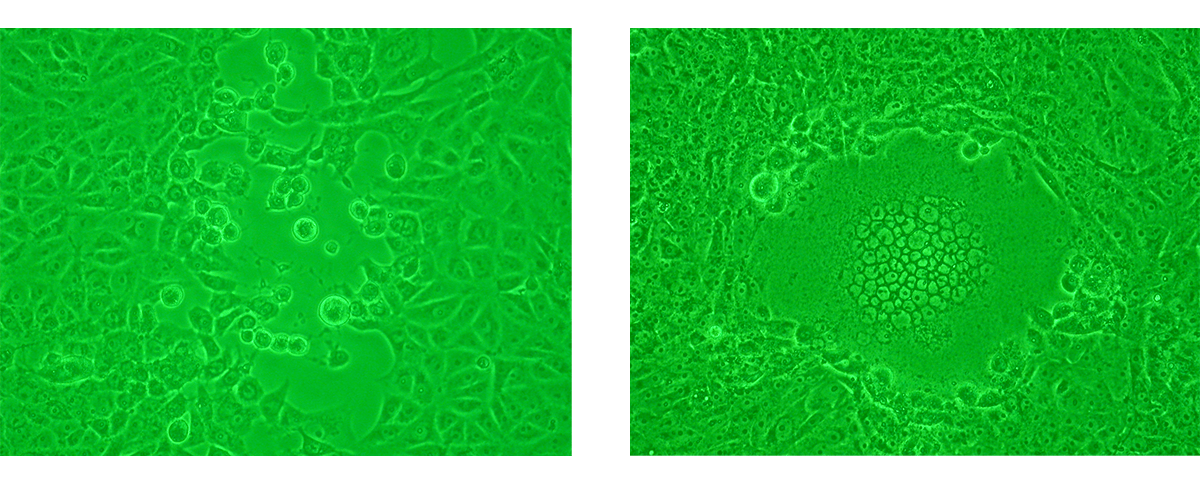
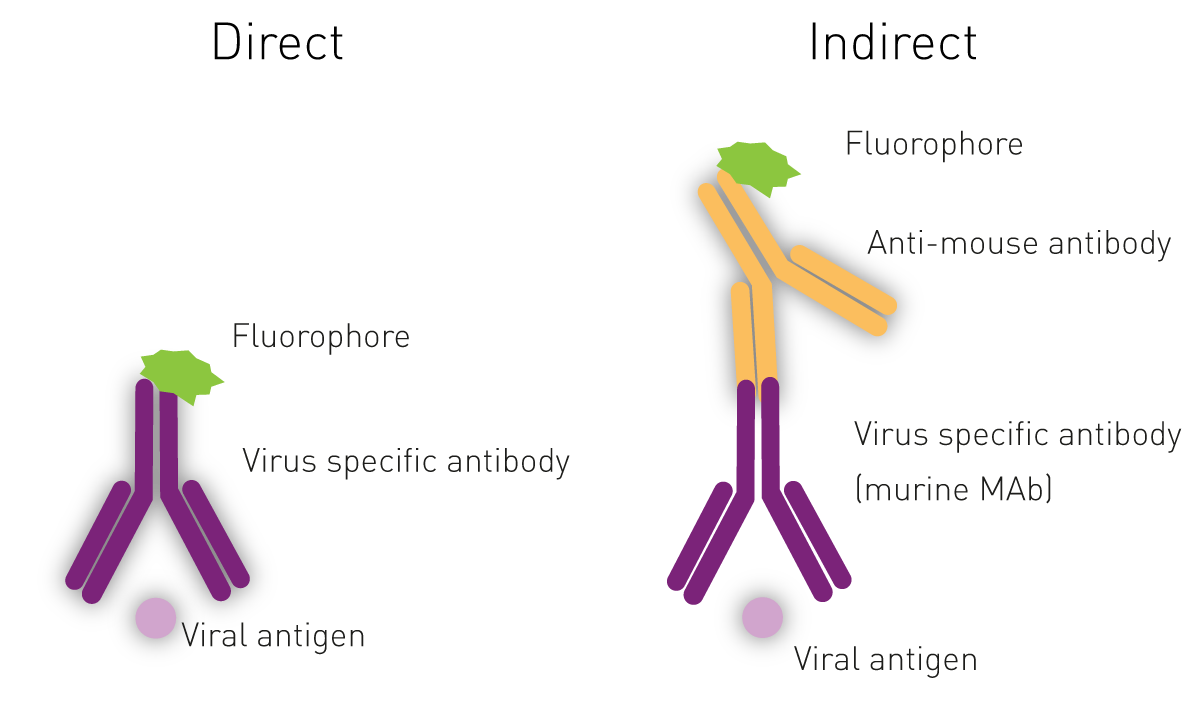
c) Fluorescence microscopy
The Immuno-Fluorescence Antibody (IFA) assay detects virus in infected cells and relies on the use of specific antibodies against a viral antigen conjugated to fluorophores. The fluorophore allows visualisation of the virus distribution by fluorescence microscopy. Whilst it is possible to predict the type of virus using the assays previously shown, confirmatory testing such as an IFA is needed for better diagnosis3,5.
d) Disadvantages of long-established microscopy virus assays
As both light and fluorescence microscopy-based virus assays rely on cell culture, they require highly trained personnel and adequate facilities. Cell culture methods are not suitable for the diagnosis of viruses which do not replicate in cell lines (norovirus, hepatitis virus) or produce CPE3,7. Additional disadvantages include the requirement of a large volume of patient sample for inoculation3, 5, 8 and long TATs. Therefore, these methods are often used more for research than for clinical diagnosis.
Modern techniques for the rapid diagnosis of viral infections
Whilst still employed, traditional virus assays have been or are being replaced by more rapid methods allowing qualitative testing for screening and surveillance, as well as confirmation of diagnosis.
a) Nucleic Acid Amplification techniques (NAATs)
Detection of viral genetic material in patients diagnoses infections early and is used increasingly due to its enhanced sensitivity, speed and reliability9. As NAAT virus assays detect and amplify specific regions of the viral genome, they confirm viral presence even before antigens and/or antibodies appear in the bloodstream.
NAATs rely on DNA/RNA extraction from the patient sample, purification and testing for quality and concentration, followed by the selection and amplification of the genetic material of interest. The Quality assessment and quantification is commonly done by absorbance measurement using an absorbance microplate reader. Absorbance is measured at 260 nm to quantify nucleic acids and at 280 nm and 230 nm to determine contaminants post purification. For this purpose, the use of a spectrometer-based microplate reader is advantageous as it can acquire the whole absorbance spectrum of a sample, determining the concentration and purity in one reading.
Alternatively, DNA and/or RNA can be quantified using different fluorescence intensity methods like Hoechst, PicoGreen, Qubit or AccuBlue. These approaches are more sensitive as they differentiate between double-stranded and single-stranded nucleic acids. Amplification of genetic material can be achieved using various methods:
Polymerase Chain Reaction (PCR) amplifies the copy number of a viral DNA region by using virus-specific primers and a thermostable DNA polymerase. A fluorescent probe binds the amplified target and emits a detectable signal. PCR cannot be used to directly detect RNA viruses and so a Reverse Transcription PCR (RT-PCR) of the RNA genome into DNA is required prior to performing the amplification reaction. Recent approaches aim to significantly reduce the time required for performing PCR procedures, as for example shown in AN370 using NextGenPCR™ and a fluorescence microplate reader to accelerate testing for SARS-CoV-2 viral RNA.
Quantitative PCR (qPCR) includes a fluorescent quantification step after each amplification cycle. Once fluorescence intensity rises above background, the detectable level proportionally corresponds to the initial number of template DNA molecules in the sample. A standard curve allows the determination of the absolute quantity of target virus DNA or transcribed RNA10.
Loop-mediated Amplification (LAMP) assay is a thermostable amplification reaction at 65ºC. Unlike PCR, it does not rely on temperature cycling, and can be hence measured in real-time in a microplate reader with periodic shaking. This is demonstrated by the example of SARS-CoV-2 in the application note LAMP assay for detecting SARS-CoV-2 RNA using an absorbance-based colorimetric readout. The assay can be colorimetric or fluorescent. Compared to PCR, this approach has speed and cost advantages. In addition, it is highly automatable, making it one of the best virus assays for rapid diagnosis11.
Next Generation Sequencing (NGS) is a generic term to describe different modern DNA/RNA sequencing technologies that offer a much quicker and less expensive approach than first generation Sanger sequencing. These methods can provide important information on novel viruses to assist in the production of vaccines and treatments against them. The accurate quantification and purity check of DNA or RNA prior to running NGS is critical to the success of the procedure.
NAATs are very useful in virus detection. However, they can neither assess its ability to replicate and infect cells, nor detect past infections or acquired immunity. To achieve these answers, quantitative infectivity and serological tests are required.
b) Quantitative virus assays
Virus quantification is crucial for research and for production of vaccines, viral antigens, viral recombinant proteins, or antiviral agents. Quantitative infectivity assays determine the capability to enter the cell and utilise its molecular replication machinery. Virus infectivity assays estimate the virus concentration and can be performed in cell culture or using egg infection. In cell culture, a titration of virus is applied to a constant number of cells and given time to invade and replicate. Infectivity is then quantified using a variety of virus assays.
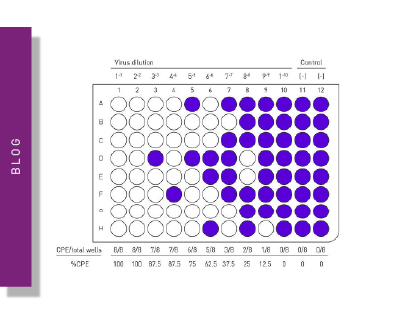
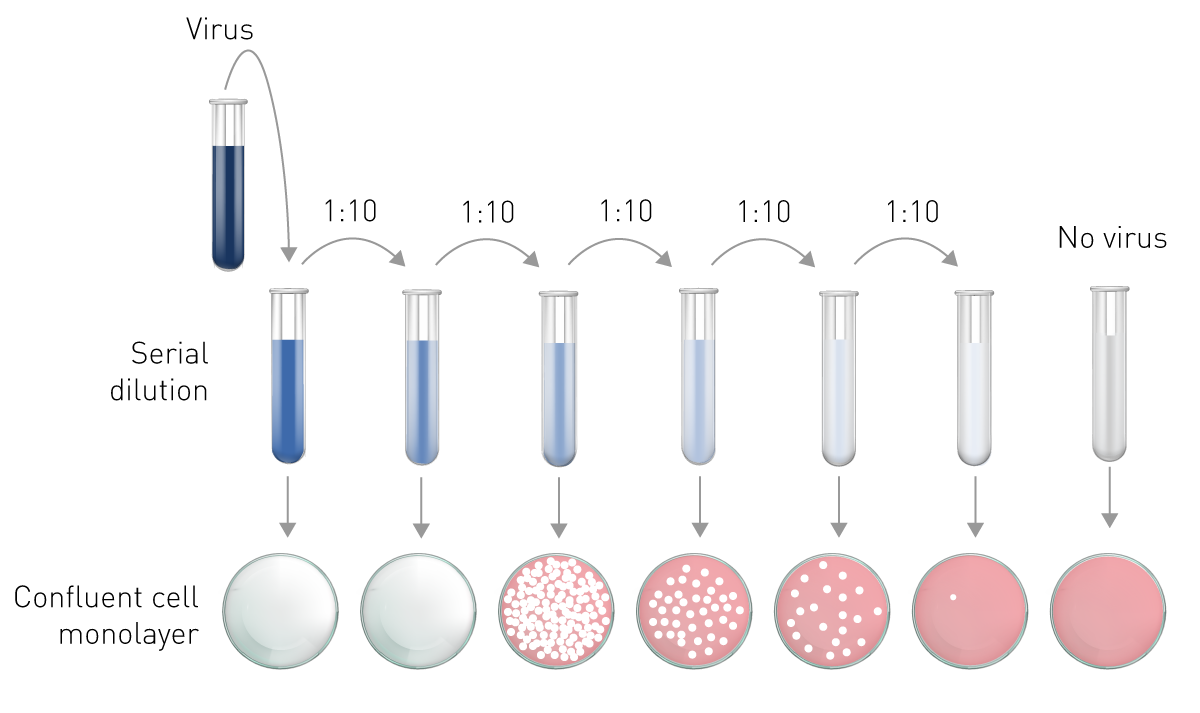
The plaque assay represents the gold standard and most used quantitative virus assay. In plaque assays, a confluent monolayer of cells is infected with unknown concentrations of a lytic virus at varying dilutions. Infected monolayers are then covered with an immobilizing overlay medium to prevent viral spread. Zones of cell death (plaques) will begin to develop as result of constrained infection and replication. Infected cells will propagate the replication-lysis-infection cycle, resulting in increasingly distinct and discrete plaques (fig. 6). Visible plaque formation can take 2–14 days, depending on the virus and host cells used. Plaques are typically counterstained by neutral red or crystal violet to be counted manually. In combination with the dilution factor used, the viral titre is calculated in terms of plaque forming units (PFU) per millilitre (PFU/ml). The pfu/ml represents the number of infective particles within the sample and assumes that each plaque formed is representative of one infective virus particle.
ELISAs essentially employ a viral antigen or antibody against the pathogen immobilised on the surface of a microplate well. This is recognised and bound respectively by viral antibodies or specific proteins of the virus present in the samples added to the well.
Specific labelled antibodies recognise the bound molecule of interest and produce a colorimetric (absorbance), chemiluminescent or fluorescent signal that is detected by a microplate reader. The signal is proportional to the amount of specific antigen or antibody in the sample.
For indirect ELISAs, detection involves a two-step process whereby a non-labelled antibody binds to the antigen or viral antibody and is then recognised by a labelled secondary antibody.
ELISAs are also used in a competitive way, measuring the concentration of an antigen by detection of signal interference. The sample antigen competes with a reference antigen for binding to a specific labelled antibody. The reference antigen is pre-coated on a multi-well plate. The sample is pre-incubated with labelled antibody and added to the wells. Depending on the amount of antigen in the sample, the free antibodies will be available to bind the reference antigen. This means the more antigen there is in the sample, the less reference antigen will be detected and the weaker the signal. This method can also be adapted to detect antibodies in samples.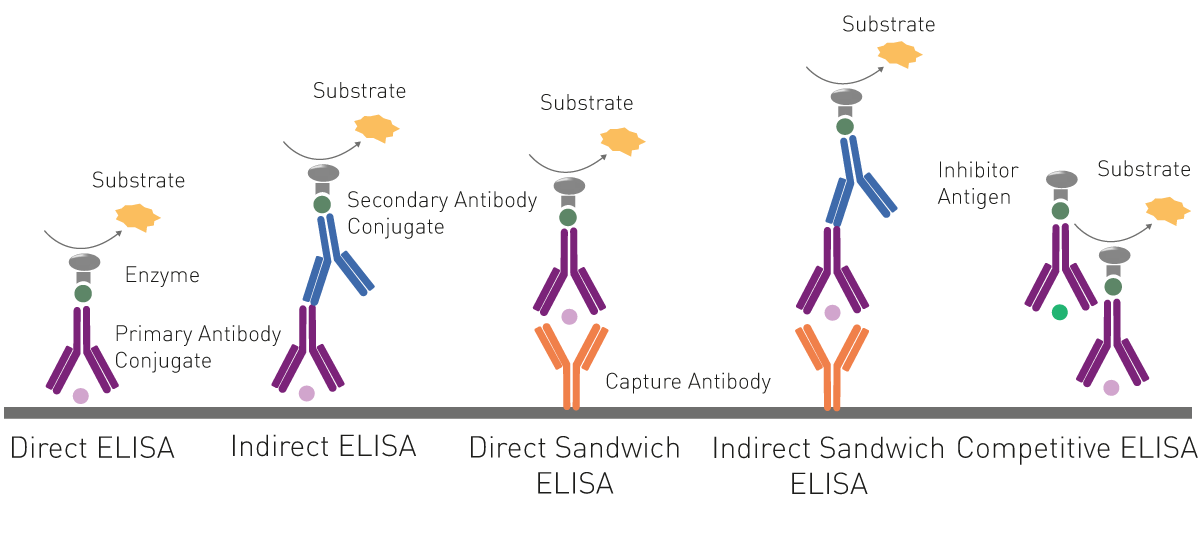
There are different types of functional assays to study how a virus works, such as how it infects cells and which mechanisms and enzymes are important for the virus. Functional methods are employed for both: basic research as well as for HTS drug screening.
The FDA suggests the use of in vitro virus assays for measuring CPE, mechanism of action and testing of resistance to novel antivirals before proceeding with clinical studies15. The basic assay principles of high throughput and research tests are similar and virus specific. Some examples are described below.

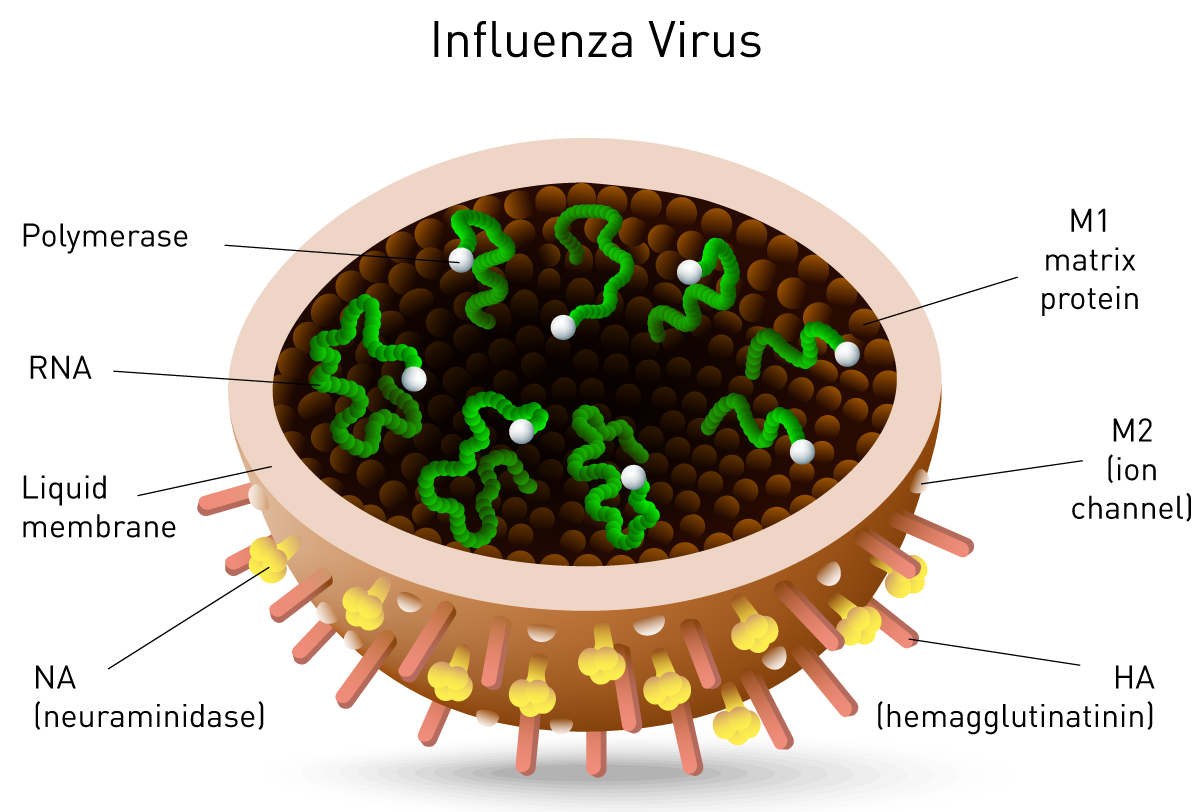
Neuraminidase assay
Neuraminidase is one of two key glycoproteins found on the surface of influenza viruses (the other being hemagglutinin, fig. 10). Neuraminidase is crucial to the release of influenza from infected cells and enables spread within the respiratory tract. Viral neuraminidase cleaves sialic acid groups from glycoproteins on the host cell surface, leading to release of virus particles from infected cells. It is therefore the target of new therapies such as the FDA-approved neuraminidase inhibitors zanamivir and oseltamivir16, 17. Its activity further reports on viral titer and potential neuraminidase inhibitor resistance.
Neuraminidase activity and inhibition is mainly measured using a synthetic substrate which releases a fluorophore, if processed by the enzyme. In this assay, the virus is mixed with the substrate and fluorescence intensity is measured on a microplate reader. The intensity is directly related to neuraminidase activity. A fluorescence-based neuraminidase assay was measured by Hong Kong-based researchers on the CLARIOstar microplate reader and revealed a neuraminidase inhibitory activity by traditional multi-herb formulations18.
A second type of neuraminidase assay employs chemiluminescence instead of fluorescence and provides higher sensitivity19. This virus assay uses a sialic acid derivative which emits light in the presence of active neuraminidase.
RNA dependent RNA Polymerase assays
Another target for RNA viruses such as such as SARS, hepatitis C and influenza is viral RNA-dependent RNA Polymerase (RdRP) (fig. 10). Viruses localized in the cytoplasm of a cell do not have access to the host’s RNA polymerase and encode their own polymerase to allow replication. The RdRP enzyme transcribes RNA from a viral RNA template, in contrast to DNA-dependent RNA polymerase that transcribes RNA from the host DNA template. RdRP enzyme activity and inhibition are important virus assays and rely on multiple RNA-dependent RNA polymerase assays.
RNA transcribed by RdRP is measured by fluorescent RNA dyes that increase their fluorescence in the presence of RNA. Recombinant RdRP or RdRP isolated from infected cells catalyses in vitro RNA synthesis. The increase in RNA is then measured using RNA dyes such as SYTO-920, Quantifluor21 or a protein-based sensor22.
Measuring nucleotide incorporation is a traditional virus assay to determine the activity of RdRP by in vitro RNA transcription. The RdRP enzyme is mixed with a biotin-labelled primer to initialize the reaction and labelled nucleoside triphosphates (NTPs). If the enzyme is active, it elongates the primer strand and incorporates the labelled NTPs. The complex of primer and newly synthesized RNA is subsequently immobilized on a streptavidin coated microplate and excessive NTPs can be washed away. Dependent on the activity of RdRP, the amount of NTPs immobilized onto the microplate differs. In the past, the NTPs were radioactively labelled. Today fluorophores are used and quantified in a fluorescence microplate reader23.
A fluorescence polarization-based (FP) assay also uses the in vitro complementation of a template RNA strand by RdRP to determine its activity. A single stranded “template” RNA is fluorescently labelled and displays a low FP value. If RdRP synthesizes the complementary strand, the molecule enlarges its mass (double-stranded) leading to an increase in FP24.

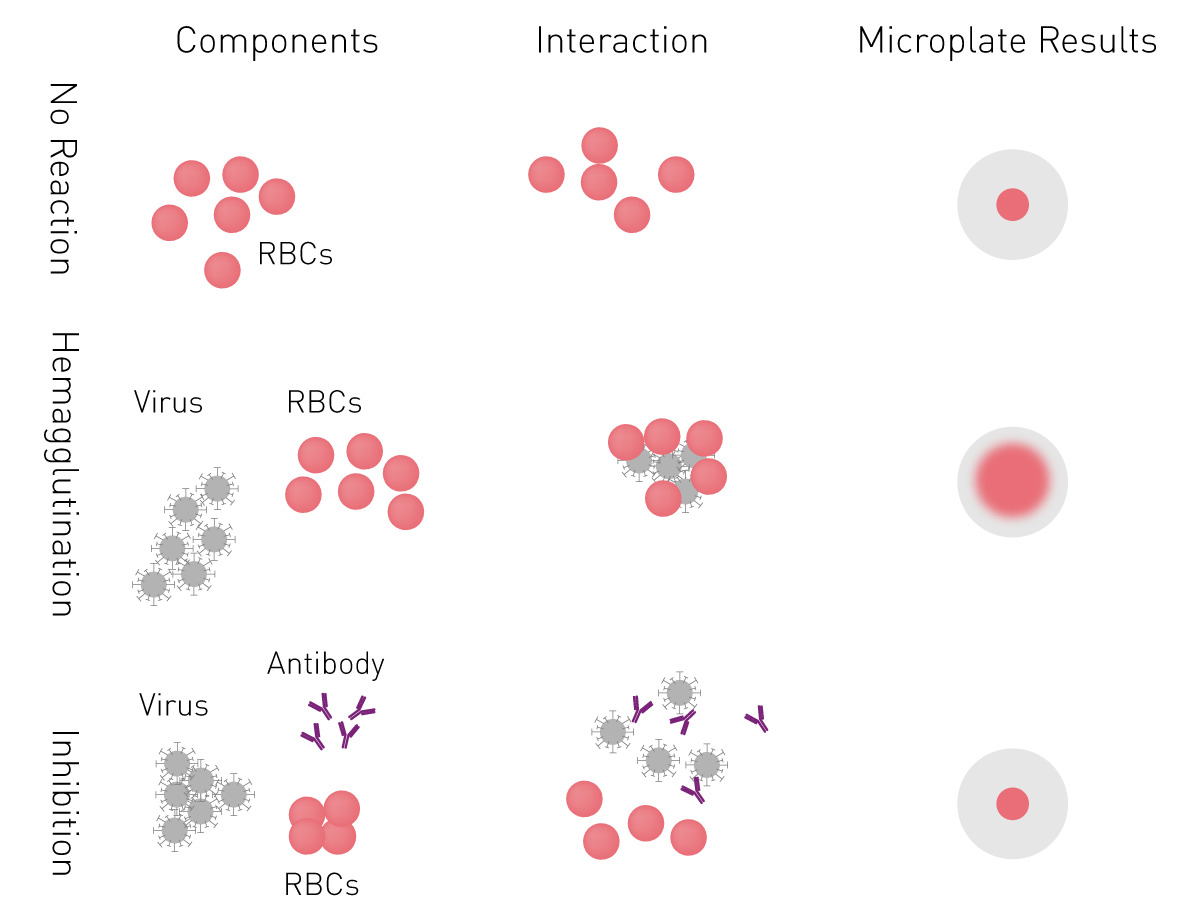
Viral neutralization assays
The effectiveness of an antibody, vaccine treatment or infection-related immunity is tested with a neutralization assay. The purpose of this virus assay is to evaluate the ability of an antibody to bind viral surface antigens that are required for a successful viral replication thus neutralizing the infection36. The neutralization assay combines incubation of the virus or pseudotypes with samples containing potentially neutralizing antibodies, e.g. sera of vaccinated humans/animals or therapeutics. This is followed by a cell-based infection assay, which provides information as to whether the virus can infect the cells despite treatment.
Neutralization assays were traditionally performed with a virus solution mixed with the neutralizing samples at different concentrations. Due to the infection risk of using the native pathogen, today, pseudotypes are used. Pseudo viruses are chimeric viruses bearing the viral glycoproteins of interest without the disease-causing phenotype. For a neutralization assay, they are engineered to express reporter genes for luciferase or GFP that are activated in the infected cell when undergoing replication. This way, the neutralization assay is easily detected in a fluorescence or luminescence microplate reader (fig. 12).
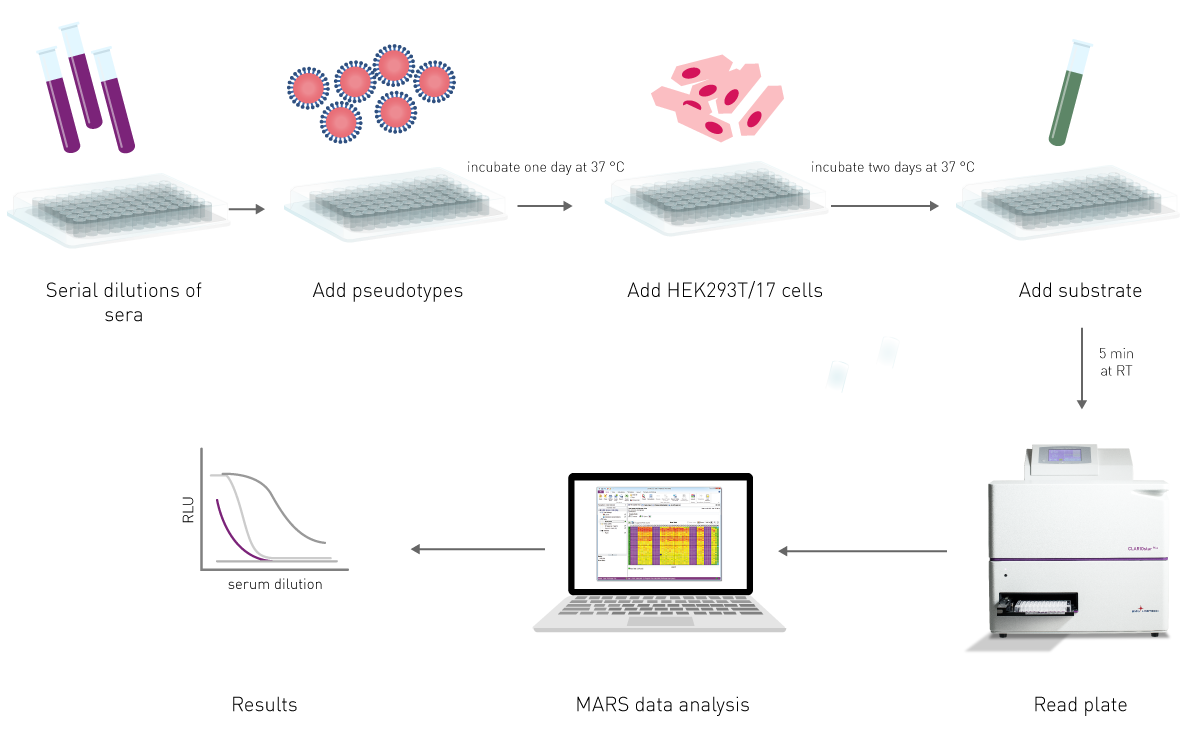
Visit our blog post “Vaccine Development: past, present and future” for more information on neutralization tests and other vaccine development assays.
Virus infectivity assays to study antiviral activity
Methods used to test the antiviral effect of compounds overlap with assays used to diagnose infections. For in vitro and high throughput assays, cell-based microplate assays are employed.
Studying the cytopathic effect of a virus in presence and absence of a compound identifies if it prevents infection. Infectivity assays to detect cytopathic effects in screening are mainly based on measuring viability with an ATP-dependent luciferase such as CellTiter-Glo® or ToxiLightTM. These assays are compatible with a 1536 well format and reagents can be added directly to cells after the appropriate incubation of cells, virus and compound. The microplate is subsequently measured on a high throughput luminescence plate reader where low signals correlate to a high cytopathic effect. In addition to a toxic effect related to infection, this assay reports on toxicity induced by the compounds themselves in control wells with compound and cells lacking the virus.
In this short talk, Ahlam Ali from Queen’s University Belfast, UK shows a screen for SARS-CoV-2 antivirals, measuring the cytopathic effect on a CLARIOstar Plus in 384-well plates. For more details see also AN 363: Viral cytopathic effects measured in a drug discovery screen.

Another infectivity assay suited to screening uses recombinant viruses that are engineered to express fluorophores or a luciferase after infection. Cells and virus are incubated together with the compounds. If infection is inhibited by one of the compounds, the fluorescent or luminescent signal is low, whereas infection is reported by high signals, as demonstrated in the application note Studying the molecular mechanism of viral replication in real time using the CLARIOstar Plus with ACU.
Binding assays for compound screens
In the context of virus research and antiviral development, binding assays screen for molecules that interact with target structures essential for the virus lifecycle. Popular target structures are: receptors on host cells used by viruses to attach and enter the cell, proteases cleaving viral precursor proteins or cell proteins, polymerases, integrases and methyltransferases required for genome replication.
Binding assays rely either on proximity assays such as resonance energy transfer, AlphaScreen assays, or the use of fluorescence polarization to track binding-induced changes in molecular weight.
a) Proximity based virus assays
Proximity assays employ donor molecules that transfer a signal to an acceptor molecule if found nearby. Two binding partners of an interaction are labelled, one with the donor molecule, the other with the acceptor molecule. The acceptor only emits a signal if binding occurs and donor and acceptor are close. Screening for binding molecules uses a competitive setup: a target molecule bearing the donor and a known target binder bearing the acceptor lead to acceptor signal emission. If a compound of a screening library displaces the acceptor-labelled molecule, acceptor signal decreases due to separation. Therefore, the signal decreases if a compound binds the viral target.
Upon excitation, the donor beads in AlphaScreen-based assays release singlet oxygen which can travel up to 200 nm and react to release light if the acceptor bead is in close proximity.
A research group of the National Institutes of Health (NIH) used this method to establish a high-throughput virus assay in 1536 well format to find inhibitors of the interaction between SARS-CoV-2 spike protein and ACE2 receptor. The viral spike protein attaches to the ACE2 receptor for cell entry and an inhibitor of the interaction is a promising therapeutic. The assay measured on a PHERAstar FSX provided an excellent assay quality with Z’ > 0.7 and identified 25 compounds out of 3400 which inhibited spike protein – ACE2 interaction25. To learn more about an AlphaLisa-based approach to study the interaction of the cellular ACE2 receptor and a viral spike protein, see our AN 357: Development of an AlphaLISA protein-protein interaction assay to screen for re-purposed drugs as targeted disruptors.
The second type of proximity assays are based on resonance energy transfer taking place between two fluorophores (FRET) or between a luciferase and a fluorophore (BRET). High throughput screens rely on simple mix and read, homogeneous assays with low background signals. The homogeneous mix and read approach applies to any resonance transfer assay.
Low background signals are observed with NanoBRETTM assays that use red shifted acceptor fluorophores, and with Time Resolved FRET (TR-FRET) that uses long lifetime donor fluorophores. TR-FRET emissions can be measured after a delay that allows autofluorescence to fade prior to signal detection. A TR-FRET based assay was used by US researchers to screen inhibitors of the interaction of HIV-1 integrase and its main co-factor. They determined a potent inhibitor which acted on integration of HIV-1 as well as on post-integration steps, therefore being regarded as a promising candidate for HIV therapy26.

b) Fluorescence polarization-based virus assays
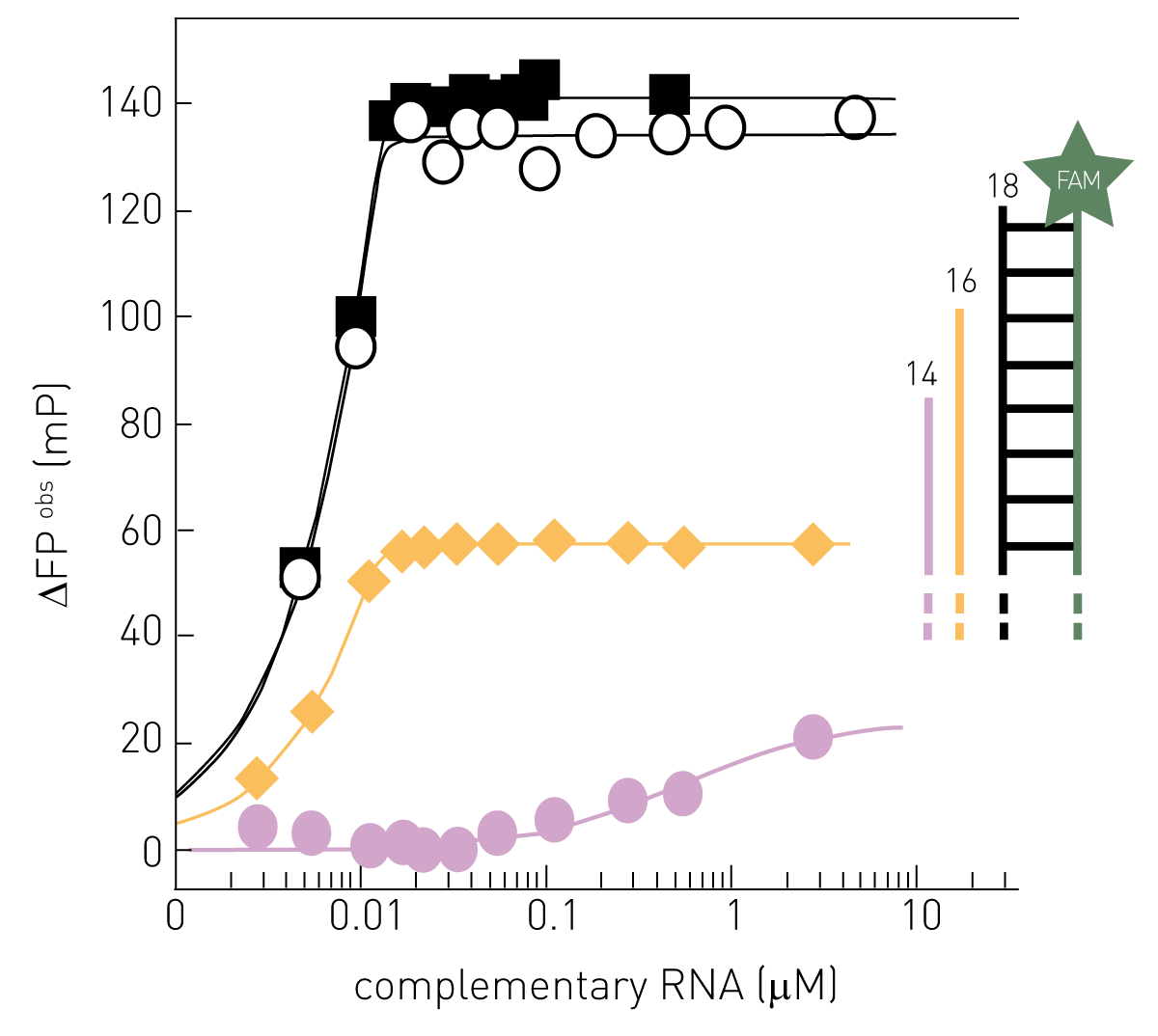
Fluorescence polarization is a cost effective and high throughput compatible method to assay the interaction of virus structures such as nucleic acids and proteins with either virus components required for replication, host proteins, or inhibitors. The fluorescence polarization of fluorophore emission increases with an increase in molecular weight of the fluorescent molecule. Therefore, binding assays based on fluorescence polarization rely on gain and loss of molecular weight of the fluorescent molecule upon binding and dissociation, respectively. Since it is easy and inexpensive to label oligonucleotides with fluorophores, FP is ideal for studying interactions between the viral genome and replication proteins. A virus assay published in “Scientific reports” employed a FAM-labelled RNA template from the Zika virus to compare how tight the polymerase subunit RdRp of NS5 and full length NS5 bind the RNA. The results helped to identify the full length NS5 protein including RdRp as well as a methyltransferase as a potential target for Zika virus therapy27
Absorbance plate reader with cuvette port
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Upgradeable single and multi-mode microplate reader series
Flexible microplate reader with simplified workflows
Gene reporter assays are sensitive and specific tools to study the regulation of gene expression. Learn about the different options available, their uses, and the benefits of running these types of assays on microplate readers.
Do you want to study viral infectivity and screen for anti-virals? This blog post highlights the advantages of TCID50 assays for the determination of viral titers in microplate format.
This blog highlights some alternative viral detection methods to PCR. Find out, which can be run on microplate readers and what advantages the readout in microplates offers.
This blog article highlights different approaches on how microplate readers can be used for the detection of viruses. Read more here.
Vaccines are powerful tools to fight viral diseases. Read more about the science behind vaccines and what we can expect from vaccine development in the future.