SPECTROstar Nano
Absorbance plate reader with cuvette port


This author profile refers to work created by Dr Andrea Krumm during her tenure at BMG LABTECH - the Microplate Reader Company, where she served as an Applications Specialist contributing scientific content, application notes, and technical expertise. Dr Krumm is no longer employed at BMG LABTECH, but her published materials remain available here for reference and archival purposes. Dr Andrea Krumm is a biotechnology specialist and product manager known for her work in analytical instrumentation and biopharmaceutical analysis. She studied biotechnology and later earned a PhD in cancer biology, focusing on topics such as DNA repair, epigenetics, and tumor biology. After completing her doctorate, she spent several years as an Applications Specialist at BMG LABTECH, where she authored application notes, conducted workshops, and supported scientific customers. In 2020, Dr Krumm joined Tosoh Bioscience GmbH as Product Manager for Analytical Columns.
Today, life expectancy in many developed countries is above 80 years of age. However a longer life expectancy also increases the chance of developing a neurodegenerative disease like Alzheimer’s or Parkinson’s disease as well as other dementias and related dementias. As these disorders progress, they become increasingly debilitating until full-time care is often required. Unfortunately, the etiology of these diseases is still not fully understood, and most available drugs only treat symptoms. With life expectancy on the rise, it is important to further our understanding of these conditions and develop new treatments. There are currently no known cures and few treatment options for most neurodegenerative disorders, and ongoing research aims to discover effective cures.
Biological assays, including aggregation, cytotoxicity, and signaling assays, are crucial to help researchers understand the underlying mechanisms of these diseases, as well as identify promising therapeutics in the early stage of drug development. Here we take a look at the importance of these assays and the need for sensitive and fast microplate readers that offer a range of detection methods.
Neurodegenerative diseases are typically identified by symptoms such as lack of motor control, cognitive deficits or mood disorders. As the name indicates, these disorders are characterized by degeneration of neurons.
Neurons are nerve cells in the spinal cord that do not replicate. If neurons are damaged, they do not recover leading to brain dysfunction and disease. Neurodegenerative diseases impact not only the brain but also the peripheral nervous system, causing nerve cell loss throughout the body and affecting the progression and symptoms of these disorders.
Although similarities are found in different neurodegenerative diseases, the root causes may vary: Huntington’s disease is caused by a gene mutation, Creutzfeldt-Jakob disease is induced by incorrectly folded protein (prion), and Parkinson's and Alzheimer's, as well as other dementias,are suspected to be caused by a combination of genetic and environmental factors. Mitochondrial dysfunction is also known to be linked to neurodegenerative diseases. Understanding the fundamental mechanisms underlying these neurodegenerative disorders is crucial for developing effective diagnostics and treatments.
A similarity shared among these neurodegenerative diseases is the way damage is induced in neurons and consequently brains. Misfolded proteins occur in all the disorders. They form amyloid fibrils, protein aggregates that lead to the formation of plaques and potentially harm neuronal cells and brain tissue. Changes in protein levels can occur prior to clinical diagnosis, and studying the course of disease progression helps researchers understand the timeline and stages of these disorders.
Researchers want to better understanding the way these proteins misfold and are interested in finding ways to interfere with their formation in the hope that this might benefit patients.
Worldwide the incidence of neurological disorders has been increasing significantly.
Symptoms like impaired movement, memory loss, mood changes, or impaired speech are common. Initially, dysfunction does not significantly impair patients and they are able to continue to lead an independent life. However, a patient’s quality of life may drastically diminish as these diseases progress and patients often end up requiring full-time care. Consequently, the social and financial burdens are considerable.
In 2021, 57 million people had dementia worldwide. Every year, there are nearly 10 million new cases. Dementia arises from a variety of diseases that affect the brain including Alzheimer’s and Parkinson’s disease. Alzheimer’s disease is the most common form of dementia and may contribute to 60-70% of cases. Dementia is currently the seventh leading cause of death and one of the major causes of disability and dependency among older people globally. Alzheimer’s Disease has an average duration of 2 to 10 years, which no doubt has an impact on the predicted economic toll in the USA of $1 trillion per annum of this disease by 2050. In 2021, around 11.8 million people worldwide had Parkinson’s disease.
Despite decades of research, there is still no cure or effective treatment for most neurodegenerative diseases, including Alzheimer's, and finding cures remains a top priority. To date, only a limited number of diagnostics and drugs are available to detect or treat neurodegenerative diseases. Most of the therapeutics only treat symptoms.Therefore, effective treatments to delay or reduce the symptoms of these debilitating diseases are needed to limit the devastating impact on individuals, families, and society
Due to the slow onset of disease and wide range of symptoms, the diagnosis of neurodegenerative disease is often only made when the disease is well underway.
Many gaps in knowledge therefore remain in understanding these disorders, including the triggers and early stages of the diseases. Therefore, research is ongoing to understand these diseases in more depth and help identify new drugs. Scientists are actively investigating new approaches to understand disease mechanisms, with a focus on the basic principles and the molecular and cell biology underlying neurodegeneration.
In recent years, new in vitro assays have become available that permit study of neurodegeneration at high throughput. These assays often utilize molecular and cell biology approaches as fundamental tools in this research.
These tools allow different experimental conditions or multiple potential drugs to be tested quickly and accurately. Several of these tools are key assays are valuable tools for analyzing disease pathway and evaluating the effects of potential drugs. Basic research in these areas is essential, as it supports efforts to develop biomarkers for early diagnosis and treatment monitoring.
Here we will look at assays testing aggregation, cytotoxicity, signaling, and protein quantity and show how they help to understand neurodegenerative diseases.
A key feature of neurodegenerative diseases is the transformation of soluble, functional proteins into insoluble, highly ordered protein aggregates termed amyloid fibrils or plaques. This transition begins with the formation of prefibrillar species (dimers, tetramers, hexamers, and other oligomers) before developing into large protein aggregate complexes. In the case of prion research, whole animal models have traditionally been used to monitor these protein aggregates through lengthy bioassays that could take up to six months. In 2012, a faster, higher throughput aggregation assay to monitor for prion seeding was developed by researchers at Rocky Mountain Laboratories in Montana called real-time quaking-induced conversion assay (RT-QuIC). Various programs and research centers collaborate to advance the development and application of these aggregation assays, supporting innovation and standardization across the field.
RT-QuIC uses fluorescence intensity to measure the aggregation of prion proteins. The fluorescent dye thioflavin T (ThT) is added to recombinant proteins. The molecule binds to beta-sheets formed during fibril formation, which induces a fluorescence increase. The method is performed in microplates and uses recombinant prion protein, tau protein, or alpha-synuclein. The microplate is intermittently shaken to permit break-up of fibrils during shaking and new fibril formation during quiescence. The process takes up to 168 h, but fibril formation is accelerated at higher temperatures. Therefore, the protein aggregation assay is often performed at 37 °C or higher. The combination of high temperature, intermittent shaking, and long-run times places high demands on the measuring instrument. The Omega series of microplate readers from BMG LABTECH provide a robust and reliable platform for this type of demanding work as outlined in the application note “Real-time quaking-induced conversion assay for prion seeding”. Features such as high-temperature incubation (up to 65 °C), an improved plate carrier, and a more robust transport system have made the Omega series of microplate readers the instruments of choice for aggregation assays.
Several programs and initiatives, such as international research funding programs and collaborative networks, support the use and further development of RT-QuIC and related assays, helping to drive progress in neurodegenerative disease research.
You can learn more about ways to optimize these types of measurements in the scientific talk Optimizing sensitivity and specificity in the RT-QuiC assay.

In 2016, a study found the RT-QuiC analysis of cerebrospinal fluid was a reliable test for sporadic Creutzfeldt-Jakob disease (McGuire et al. 2016). Eleven centers based in Europe, Asia, and Australia analyzed samples of human cerebrospinal fluid using RT-QuiC and BMG LABTECH plate readers (a twelfth center used a microplate reader from another manufacturer). The centers diagnosed Creutzfeldt-Jakob disease with a specificity of 100 %.
In the application note “Novel aggregation-specific fluorogen monitors prefibrillar protein aggregation by fluorescence polarisation (FP)” a novel fluorophore bis(triphenyl phosphonium) tetraphenylethene (TPE-TPP) was used to investigate the onset of protein aggregation. The TPE-TPP dye has superior characteristics to the ThT dye as it binds more efficiently. Furthermore, the monitoring of its fluorescence polarization (FP) reports on early stages (dimers, tetramers, etc.) of aggregation. FP measures the rotation of molecules in solution. Small molecules move fast and depolarize the emission whereas large molecules move more slowly and retain emission polarization. The binding of TPE-TPP to (pre-)amyloid structures slows down its rotation leading to measurable changes in FP. The assay was developed by a research group in Australia that used a CLARIOstar® microplate reader to perform the FP measurements.
Besides protein aggregation, neuronal cell death is a hallmark of neurodegeneration. While neurons do not undergo cell death in healthy adults, they do die during neurodegenerative diseases which leads to loss of brain function. The neurons die via mechanisms of programmed cell death which can be induced by oxidative damage of mitochondria or DNA, membrane damage by protein aggregates, and other processes. Studying these mechanisms in both animal models and humans is crucial, as research involving humans provides insights that non-human models alone cannot capture.
Apoptosis, necrosis, and autophagy have been reported as mechanisms of programmed cell death in neuronal cells. Researchers want to understand better the causes and mechanisms of neuronal cell death to help find new drugs to treat neurodegenerative diseases such as Huntington's disease, amyotrophic lateral sclerosis (ALS), and Alzheimer's disease. Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a major focus in neurodegenerative disease research, with efforts aimed at understanding disease mechanisms and developing therapeutic strategies.
Many assays are available to study cell death and can be used to investigate changes in neurons. You can read more about cytotoxicity assays in our blog “Cytotoxicity – These assays tell you what your cells don’t like”.
The most used cytotoxicity assays are based on tetrazolium salts. Their reduction by viable cells leads to an absorbance shift that is read by microplate readers and indicates metabolic activity. Researchers from the National Medical Research Center, Moscow, Russia used this type of assay to prove that insulin limits excitotoxicity in cortical neurons, an effect inducing cell death in neurons that is linked to neurodegenerative diseases (Krasil’nikova et al. 2019).
ATP-dependent luciferase viability assays are popular because of their simplicity and sensitivity. They measure the ATP content of lysed cells by an ATP-dependent luciferase. The light output measured by a microplate reader in this type of assay is directly related to cell number. The assay helped researchers to reveal a cytotoxic effect of mononamine oxidase (MAO) activity in a mouse cell model of Huntington disease (Ooi et al. 2015), and similar approaches are used in ALS research to identify disease mechanisms and potential treatments.
Recently cell death assays have been developed not only report on toxicity at one-time point but to monitor cytotoxic effects over time. The RealTime-Glo™ viability assay produces light in the presence of viable cells. If cells are cultured in a controlled environment (37 °C, 5 % CO2) by an atmospheric control unit, RealTime-Glo™ viability assays can be used to monitor viability over days. The Atmospheric Control Unit (ACU) for the CLARIOstar and VANTAstar provides versatility in long-term cell-based assays. Using the assay on a CLARIOstar microplate reader, a protective effect of a specific fusion protein against neurotoxins was studied (Paliga et al. 2019). These advancements support the next generation of research tools and methods for studying neurodegenerative diseases, including ALS, Alzheimer's, Parkinson's and other disorders.
An early event of neurodegeneration is impaired signaling of neurons. Altered signaling processes include the control of synaptic transmission by Ca2+ signaling , energy metabolism, and cell survival or signal transmission via receptors. These disruptions are a major focus for research into understanding neuronal signaling in neurodegenerative disease research.
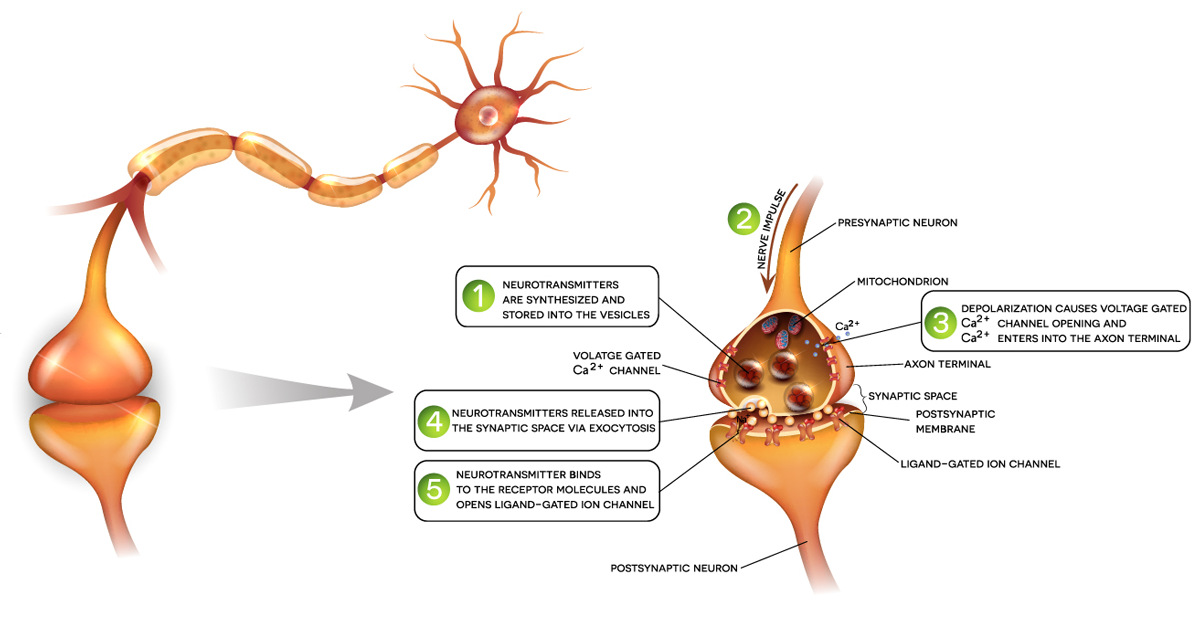
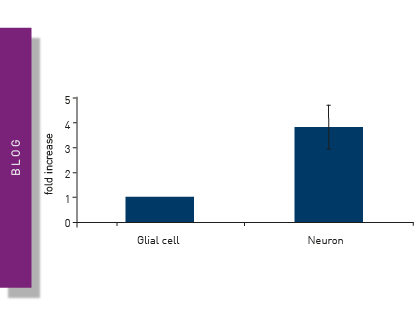
Ca2+ signaling in neurons is vital for their cellular function as information transmitters. Intracellular Ca2+ changes can be monitored by fluorescent dyes such as Fura-2, Fluo-8, or Cal- 520. The application note “Monitoring intracellular calcium using fluorescent dyes in a mid-throughput assay” compares the different assays monitoring Ca2+. This study provides an example of how Ca2+ assays are beneficial for neurodegenerative diseases research. Transient levels of Ca2+ were measured in real time in human endothelial cells. (Ferguson et al. 2016). Fura-2 intracellular Ca2+ measurements showed the stimulation of differentiated cells with KCl, while non-differentiated cells did not increase intracellular Ca2+ levels upon KCl treatment. Such attributes make these dyes and assays useful for mid-/high-throughput analysis of intracellular Ca2+ levels in living cells. These findings are clinically relevant, as they contribute to advances in medicine by informing the development of new treatments for neurodegenerative diseases.
Several proteins show altered expression during different neurodegenerative diseases. Tau protein is implicated in Alzheimer's disease, the TREM2 receptor (Triggering Receptor Expressed on Myeloid cells 2 receptor) correlates with the risk of developing Alzheimer’s disease, and Brain-Derived Neurotrophic Factor (BDNF) is decreased in Alzheimer’s disease. For Parkinson’s disease, increased levels of neuroinflammation are linked to changes in the levels of cytokines such as IL6, IL β1, and TNF α.
A popular method to quantify specific proteins in solution is ELISA. Our application note “Fast and accurate detection of Alzheimer’s Disease targets with SimpleStep ELISA® kits and SPECTROstar® Nano” explains the SimpleStep ELISA principle and shows how it was used to quantify proteins related to neurodegeneration. SimpleStep ELISA streamlines conventional ELISA assays by making it possible to run them in a semi-homogeneous format that results in a 90-minute single-wash protocol.
A research group based in Sydney, Australia, used a TNF-alpha ELISA to study the effect of chronic microglial activation on the inflammatory marker. They found pro-inflammatory TNF-alpha to be elevated at all ages of a mouse model with chronic microglial activation, a feature of neurodegenerative diseases (Gyengesi et al. 2019). Recently, phospho-tau sites Thr181 and Thr217 have shown potential as biomarkers of disease. When comparing site specific phospho-tau in normal and Alzheimer’s disease brains, these biomarkers were confirmed to functionally distinguish between healthy and diseased tissue.
These studies demonstrate high-throughput methods used in the research of neurodegenerative diseases. As they require various detection modes, the use of multimode plate readers is recommended.
For the performance of aggregation assays, it is essential that the analysis instrumentation can shake and incubate microplates over long periods of time (up to 7 days). BMG LABTECH is a technology leader in the microplate field. The multimodal plate readers include the CLARIOstar® Plus and Omega series, which have a dedicated plate carrier and provide robustness in long-term shaking aggregation measurements.
For further information on how BMG LABTECH plate readers help neurodegeneration diseases research have a look at our neuroscience research area.
Absorbance plate reader with cuvette port
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Upgradeable single and multi-mode microplate reader series
Flexible microplate reader with simplified workflows
Neurodegenerative disease ultimately leads to the death of neurons as neuronal functions deteriorate. Find out how microplate readers can be used to study neuronal cell death and its link to neurodegenerative disease.
The disruption of mitochondrial function is linked to neurodegenerative diseases. Find out how microplate readers advance research into mitochondrial dysfunction and neurodegenerative diseases.
The way proteins misfold and aggregate is linked to many neurodegenerative diseases. Find out how microplate readers can help advance research into protein misfolding.
Alpha-synuclein is a key protein involved in neurodegenerative diseases like Parkinson’s. Find out how microplate readers can help advance alpha-synuclein research.
Amyloids are thought to play a crucial role in neurodegeneration. Find out how microplate readers help advance amyloid research.
The tau protein plays a role in many neurological diseases and disorders. Find out about neuronal toxicity induced by tau and how microplate readers can aid tau research.