SPECTROstar Nano
Absorbance plate reader with cuvette port

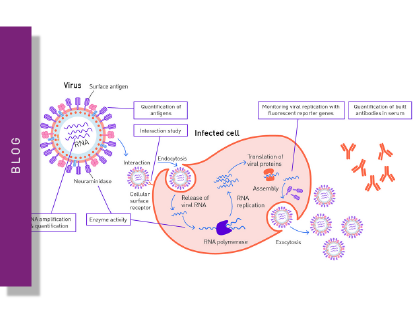
Viruses consist of nucleic acids surrounded by a protein coat and sometimes a membranous envelope. Since viruses only encode a limited number of proteins, they can only reproduce parasitically in host cells. For this purpose, they exploit cellular machineries and pathways of their host to replicate and spread. After the initial attachment to a host cell, viruses penetrate the membrane and release their viral genome into the cytoplasm. Subsequently, the machinery of the host cell is diverted to replicate the viral genome and translate it to viral proteins. After the assembly of the produced components to virions, these new viruses can be released and may infect additional cells.
Microplate readers are an indispensable tool in virology research. Functional methods rely heavily on microplate-based assays and are employed in both, basic life science research labs as well as in drug screening facilities. In fact, once the functioning mechanism of a virus is characterised and potentially druggable target proteins are identified, functional assays need to be scaled up to screen for antiviral molecules that inhibit those very mechanisms. Pharmaceutical regulatory agencies suggest the use of in vitro assays to determine cytopathic effects, resistance to novel antivirals, and mechanisms of action of a virus before moving to clinical studies 1. Microplate readers deliver robust and reproducible results and can easily be integrated into automated systems, which makes them ideal platforms for the setup of high-throughput facilities. During the Corona pandemic, in particular, methods based on serological testing or the amplification of nucleic acids have gained prominence. Apart from this, however, there are also many alternative methods, some of which have been in use for many years. The most relevant microplate-based assays utilised to detect viruses are described below.
Modern medicine combats viral infections by vaccination and drug treatment. While vaccinations confer artificially acquired immunity to a specific virus, antiviral drug treatments typically target and reduce virus entry into cells and viral replication. Thus, the effectiveness of vaccines and drugs can be assessed by determining remaining or increasing virus numbers. Besides the direct detection of viruses, many virus assays employ host cells to study viral biology. Among these, functional assays are thought to provide information on how a particular virus infects a cell. They aim at understanding the host-virus interactions, clarifying the molecular mechanisms behind the infection.
Neuraminidase assay
Neuraminidase is a key viral glycoprotein involved in the virion release from infected cells. It is found on the surface of influenza viruses and cleaves sialic acid groups from glycoproteins on the host cell surface. Its role is crucial for viral spread in the respiratory tract.
Neuraminidase activity and inhibition can be efficiently measured on plate readers using a synthetic quenched substrate. After processing by the enzyme, the substrate releases a fluorophore. Fluorescence intensity is detected on a fluorescence microplate reader and correlates with neuraminidase activity 2. Alternatively, a chemiluminescence substrate may be used, purportedly providing higher sensitivity than fluorescence 3. New therapies e. g. based on zanamivir and oseltamivir target neuraminidase 4,5.
Other enzymatic targets include for instance NSP14, an exonuclease of the coronavirus family that is currently investigated as anti-viral drug target, as discussed by Joao Pisco in this interview:

Neutralization and infectivity assays
Infectivity assays determine and quantify the activity of a virus. They can be used to investigate the viral capability to enter the host cell and to use its molecular replication machinery. The determination of antiviral drug effectiveness is thereby partially based on the same methods used to diagnose infections. Live cell-based microplate assays are generally employed for both low- and high- throughput assays.
Traditionally, neutralization assays have been performed using virus solutions mixed with potential neutralizing samples at different concentrations. The use of native pathogens exposed the operator to elevated infection risk. While virus neutralization assays are still the gold standard to assess the effectiveness of antiviral compounds or antibodies, nowadays, native pathogens have been replaced by pseudo-viruses or pseudo-types in these applications. Chimeric viruses generally only encode the viral proteins of interest without the disease-causing genes.
Neutralization refers to the binding of an antibody or compound and subsequent neutralization of viral surface proteins which are essential for host cell entry. For this purpose, the virus (or pseudo-virus) is incubated with potentially neutralizing compounds or antibodies. Incubation is followed by a cell-based infection assay. This provides information on whether a treatment can block viral infection of host cells. Infection of cells is monitored based on the expression of fluorescent or luminescent reporter genes, which were previously introduced into the genome of the pseudovirus (fig. 1A+B). This modification additionally enables monitoring infection and neutralization in high-throughput and in real-time by employing a microplate reader with gas control (fig. 1C) 6. See also: AN 358: Antiviral assay based on the expression of fluorescent proteins by the viruses, AN 353: Studying the molecular mechanism of viral replication in real-time using the CLARIOstar Plus with ACU and AN 172: Dual-luciferase assay to assess the replication of the hepatitis C virus subgenomic replicon.
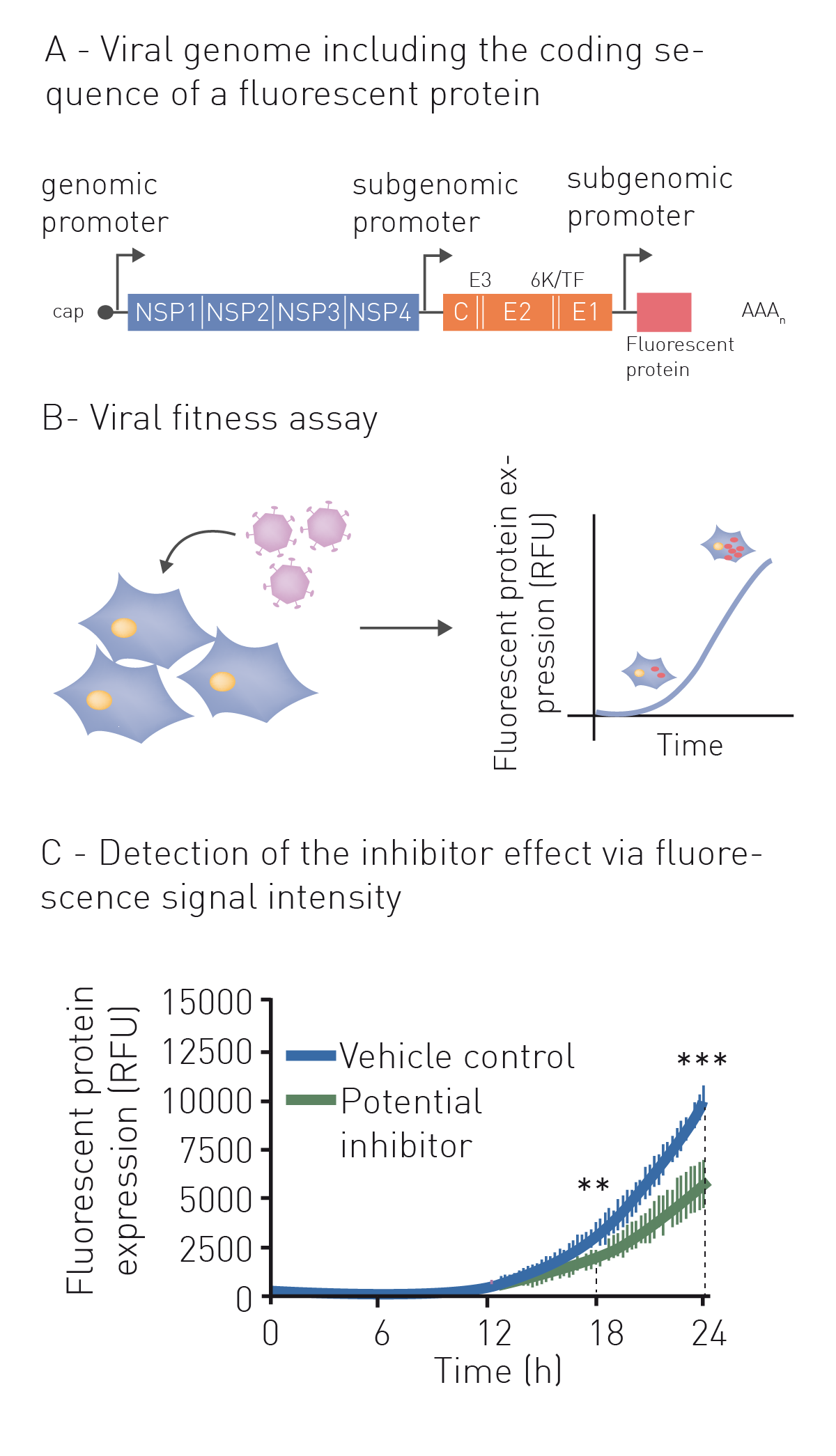 Indirect neutralization assays based on host viability
Indirect neutralization assays based on host viability
Next to the direct detection of viral replication, the study of cytopathic effects of a virus can be used as an indirect measure to identify the antiviral capabilities of test compounds or to evaluate the antibacterial properties of bacteriophages. Culture of cells and evaluation of cell viability can be performed simultaneously in a microplate reader where low signals correlate to a high cytopathic effect. Screening assays to detect cytopathic effects are mainly based on the measurement of cell viability with an ATP-dependent luciferase or on the use of cell impermeable dyes to analyse membrane integrity (e.g., Promega´s CellToxTM Green), and are compatible with microplate formats up to 1536 wells. Reagents are added directly to cells after the appropriate incubation with virus and compound. In addition to infection-derived cell death, this assay also reports on compound-induced cell toxicity in control wells which include compound and cells - but no virus.
In this short talk, Ahlam Ali from Queen’s University Belfast, UK shows a screen for SARS-CoV-2 antivirals, measuring the cytopathic effect on a CLARIOstar Plus in 384-well plates. For more details see also AN 363: Viral cytopathic effects measured in a drug discovery screen.

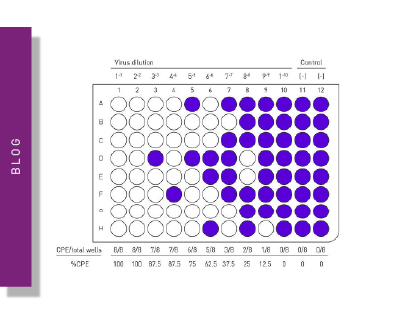
Tissue Culture Infective Dose (TCID50) quantifies the amount of virus required to produce cytopathic effects in 50% of inoculated cells. This value can also be determined with a microplate reader. While the number of host cells is kept constant, a titration series of the virus is applied. TCID50 results are expressed as 50% infectious dose (ID50) per millilitre (ID50/ml). TCID50 is regularly applied for virus quantification and is crucial in the development and testing of vaccines and antivirals, as well as the production of viral antigens and viral recombinant proteins.
Assays based on RNA dependent RNA Polymerase
RNA-dependent RNA Polymerases (RdRP), found in viruses such as SARS-CoV-2 and influenza, are employed by the pathogens to transcribe RNA from viral RNA templates. Viruses localized in the cytoplasm of a cell cannot access the host RNA polymerase and therefore encode their own polymerase to promote replication. This enzyme is an important target to analyse viral activity and screen for potential inhibitors.
Traditionally, RdRP activity was determined in vitro by the measurement of radioactive nucleotides, which were incorporated during transcription. Today, the use of fluorophores and microplate readers enables a more efficient and cost-effective quantification 7. Instead of radioactive, fluorescently labelled nucleoside triphosphates (NTPs) are used. If RdRPs are active, they elongate a biotin labelled primer strand, which is subsequently immobilized on a streptavidin-coated microplate. After excessive NTPs are washed away, the amount of immobilized NTPs can be quantified by the plate reader and correlates with the activity of the RdRP. Alternatively, fluorescent dyes that increase their emission intensity in the presence of RNA can equally be used to quantify multiplied RNA at a microplate reader.
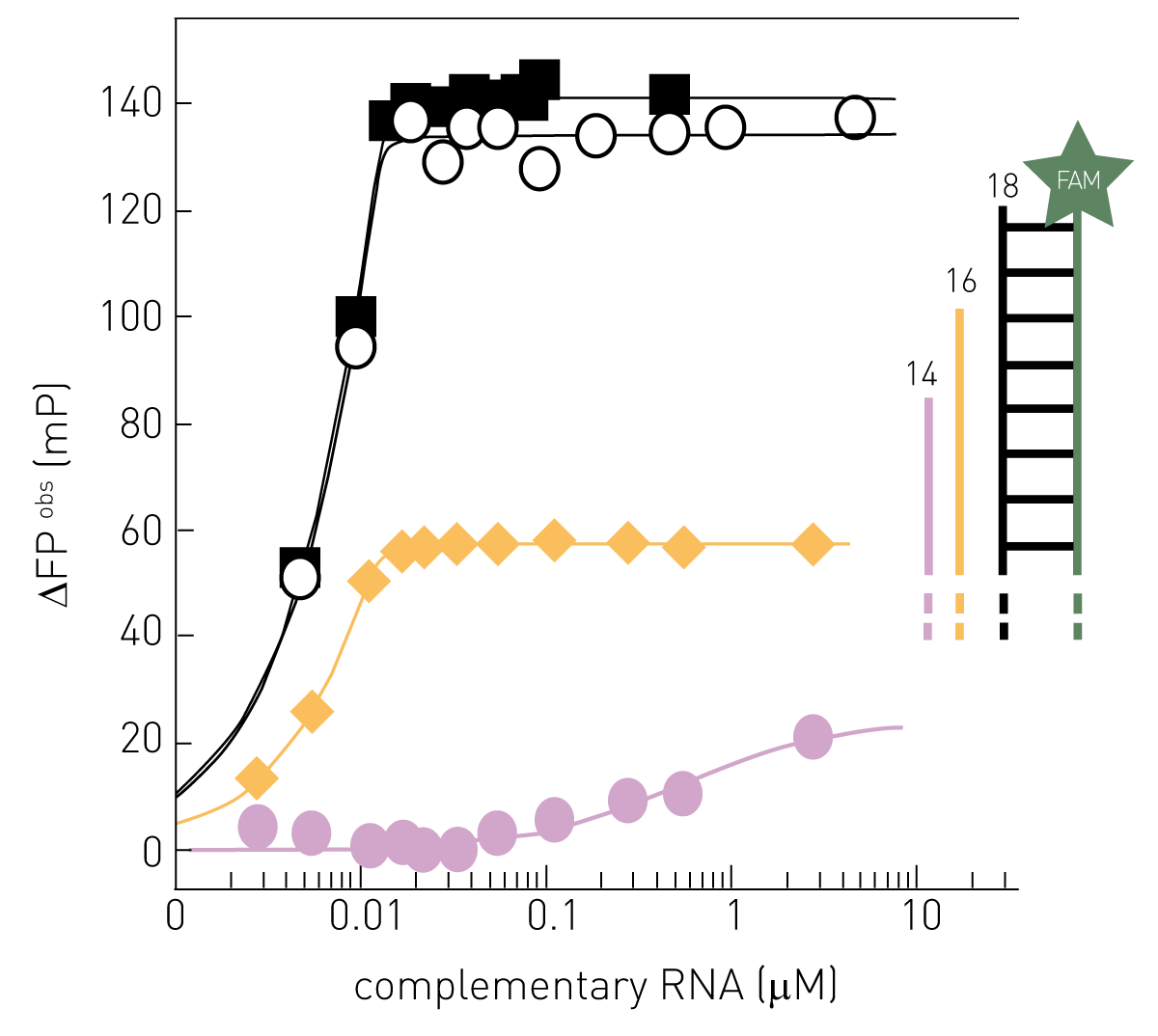
Alternatively, fluorescence polarisation can be used to determine RdRP activity by its complementation of a template RNA strand. If fluorescently labelled single-strand RNA is not elongated, it shows a low polarization value. The RNA molecule increases its size, if the complementary strand is synthesised by the RdRP, so does the fluorescence polarization signal (fig. 2). 8 Like fluorescence intensity, this method can efficiently be run on a plate reader with fluorescence polarization detection. See also: AN326: Fluorescence polarization-based RNA synthesis assay
Compound screening based on interaction assays
Interaction assays are of utmost importance for the screening of potential antiviral drugs. Viruses differ in the way they interact with cells and may use different enzymes and mechanisms for this purpose. Therefore, effective drugs to inhibit the individual steps of viral interaction can also differ greatly depending on the pathogen. Interaction assays are used to screen for molecules that interact with and inhibit structures essential for the lifecycle of a virus. Common targets include host cell receptors used by viruses to enter the cell, proteases cleaving viral precursors or cellular proteins, as well as polymerases, integrases, and methyltransferases involved in viral genome replication.
The interaction of two binding partners requires them to be in close proximity to each other. Methods based on resonance energy transfer (FRET, BRET, and TR-FRET) and AlphaScreen® depend on the proximity of two fluorophores respectively luminophores, which are bound to target structures. Therefore, their assay principle is perfectly suited to monitor the interaction of potential binding partners.
In these methods, the proximity of the target structures is assessed by Förster’s resonance energy transfer taking place between two fluorophores (FRET), a luciferase and a fluorophore (BRET), or between a fluorophore with long emission half-life (lanthanide) and a fluorophore (TR-FRET). Regardless of the method, energy is transferred from a donor to an acceptor chromophore, provided the binding partners are in proximity. For this purpose, two binding partners of interest are labelled with either the donor or the acceptor chromophore. Since the acceptor only emits a signal if the binding between the two molecules of interest occurs, it can be used as means to investigate their interaction.
To avoid the need to label a large number of potential inhibitors prior to screening, a competitive setup is used in general. Here, instead of the potential inhibitors, the corresponding target structures are labelled with fluorophores or luminophores: A target bearing the donor and a known target binder bearing the acceptor molecule. If a compound displaces the acceptor-labelled molecule, the acceptor signal decreases due to separation, signaling binding inhibition.
FRET-based methods offer a range of advantages – especially for high-throughput screening. Since donor emission is monitored in relation to the acceptor emission, data are internally normalised and the influence of the number of molecules present in a sample is eliminated by this ratiometric measurement. This way, interferences from variable assay conditions can be minimized. Particularly low background noise is observed in assays that use long lifetime fluorophores (TR-FRET) or luminophores (BRET) as donors. Furthermore, many FRET-based assays can be performed as simple mix-and-read, homogenous assays. This means that previously unavoidable intermediate steps can be eliminated which makes the transfer to a high-throughput process much easier. Recent achievements like Simultaneous Dual Emission, Alpha Technology Lasers or TRF Lasers and Decay Curve Monitoring additionally enable an increase in sensitivity and measuring speed resulting in significant benefits for high-throughput campaigns.
Many success stories in antiviral drug screening are based on the use of ratiometric measurements. For instance, a TR-FRET-based assay was used to screen for inhibitors of the interaction between the HIV-1 integrase and its main co-factor (LEDGF). Using this method, a strong inhibitor being regarded as a promising candidate for HIV therapy could be identified. 9 In a different approach, AlphaScreen has been used to investigate inhibitors of the interaction between the SARS-CoV-2 spike protein and the ACE2 receptor. Engagement of the ACE2 receptor by the spike protein allows entry into host cells and an inhibitor of this interaction could be a promising therapeutic. This high-throughput assay was established in 1536 well plates and provided an excellent assay quality with Z’ > 0.7 and identified 25 compounds out of a library of 3400 which inhibited the spike protein – ACE2 interaction. 10 To learn more about an AlphaLisa-based approach to study the interaction of the cellular ACE2 receptor and a viral spike protein, see our AN 357: Development of an AlphaLISA protein-protein interaction assay to screen for re-purposed drugs as targeted disruptors.
Next to AlphaScreen and FRET-based assays, fluorescence polarization (FP) is a cost-effective and high-throughput compatible method to screen for molecular interactions. If the molecular size of a binding complex increases, so does the polarization of the emitted light of an attached fluorophore. Accordingly, FP-based interaction assays rely on the binding- or dissociation-dependent gain or loss of molecular weight of the complex, respectively. FP-based assays can be used to monitor binding events between nucleic acids or proteins and virus components required for replication, host proteins, or inhibitors. Thus, FP is ideally suited for studying interactions between the viral genome and replication proteins in particular. For this purpose, oligonucleotides representing specific regions of their viral genome are labelled fluorescently, an approach that can be implemented easily and cost-effectively. A FAM-labelled RNA template from the Zika virus was for instance used to analyse the binding of RdRP to RNA. Based on these results, a methyltransferase was identified as a potential Zika virus therapeutic target11.
Modern microplate readers offer increased performance and sensitivity for interaction assays and can deliver robust results with minimal variability and larger assay windows. The advantages are particularly evident when using red fluorophores. The use of red fluorophores can be advantageous because this way, unspecific background signals in the green and blue range can be excluded. Low photon yield of red FP fluorophores has previously been a problem which, however, does no longer represent a limitation due to technical improvements in signal detection.
Besides the need for a profound understanding of SARS-CoV-2 biology and, building on this, for the rapid development of therapies, the COVID-19 pandemic put significant efforts in the development of diagnostic methods to identify infected individuals. Especially during an active pandemic outbreak, viral detection methods are needed to monitor current infections and their distribution in the population. Tight monitoring of the incidence level, including the identification of underlying subtypes, is necessary to control and limit virus propagation via effective disease management 12. Next to enduring diseases, this also includes past and asymptomatic infections. Precise diagnostic tools help to prevent transmission and support appropriate therapies and treatment monitoring 13.
Available diagnostic assays vary in efficacy, approach, and cost. Factors to consider when selecting an assay include ease of use, accuracy, precision and specificity, throughput, and turn-around time (the interval time between sample registration and result reporting) 14.
Traditionally, methods for the identification of viral infections focussed mainly on visual detection and counting of virions by microscopy techniques. As these methods require a relatively large volume of patient samples, are subject to long turn-around times, and are dependent on adequate equipment and highly trained personnel, they are now more often adopted for research rather than for diagnostic purposes. 13, 15, 16 Today, microscopy techniques have been replaced by diagnostic methods, which allow for faster qualitative testing, screening, and surveillance of virus infections. These rely mainly on the detection of the viral genome within patient samples or on serological tests.
Diagnosis of acute infections by nucleic acid amplification
Amplification of viral genetic material, followed by its detection represents a sensitive, fast, and reliable method to diagnose an infection in patient samples 17. As patient-derived viral nucleic acids are amplified exponentially during the procedure, the method oftentimes allows confirmation infections even before symptoms are evident, or antigens and/or antibodies appear in the bloodstream. Common methods include Polymerase Chain Reaction (PCR) and quantitative PCR (qPCR). Detection of the amplified nucleic acids can either be performed directly on an absorbance microplate reader or on a fluorescence microplate reader after appropriate staining with fluorescent dyes like Picogreen or Qubit. Both, PCR and qPCR, are based on a series of cycles of temperature changes, each resulting in a doubling of the previously present nucleic acid molecules. While regular PCR delivers a qualitative result, qPCR additionally enables to quantitate the initial amount of nucleic acid. Recent approaches aim to significantly reduce the time required for performing PCR procedures, as for example shown in AN370 using NextGenPCR™ and a fluorescence microplate reader to accelerate testing for SARS-CoV-2 viral RNA.
A relatively new approach that is increasing in popularity is the Loop-mediated Amplification (LAMP) assay. Unlike classical PCR approaches, LAMP does not require cyclic temperature changes and can be run at a constant temperature of 65°C. The detection of the amplified nucleic acid can be either absorbance- or fluorescence-based and can easily be measured in real-time at a microplate reader with periodic shaking. This is demonstrated by the example of SARS-CoV-2 in the application note LAMP assay for detecting SARS-CoV-2 RNA using an absorbance-based colorimetric readout. LAMP has already been successfully applied to SARS-CoV-2 testing18. In comparison to regular PCR-based methods, it bears cost and speed advantages, is easy to use, and therefore highly automatable, making it a perfect assay for rapid virus diagnosis.
Diagnosis of past and acute infections with antibody tests
In order to obtain a holistic impression of the infection incidence in a population, it is not enough to test for acute infections. Any existing immunity from previous infections must also be considered. A shortcoming of genetic amplification techniques is that they cannot report on acquired immunity or past infections. To get these answers, serological tests are required.
With Enzyme-Linked Immunosorbent assays (ELISA) antibodies against a virus (fig. 3) or viral antigens can be determined qualitatively and quantitatively in serological samples. During an infection, antibodies are produced by blood cells and can persist for up to several decades. The antibody titre may vary depending on the individual, strength of infection, and the time that has passed since the infection. ELISAs allow to determine patient immunity upon exposure, reinfection, or reactivation and are invaluable for studying the epidemiology of the disease. The assay produces a colorimetric, fluorescent, or chemiluminescent output that is detected by a microplate reader. The signal is proportional to the amount of specific antigen or antibody in the sample. Besides the recognition of antibodies, immunoassays are also helpful to understand the immunology behind the viral infection, detecting and quantifying for instance different cytokines. Furthermore, ELISAs can be applied to detect specific antigens of a virus, such as a spike protein or the nucleocapsid protein.
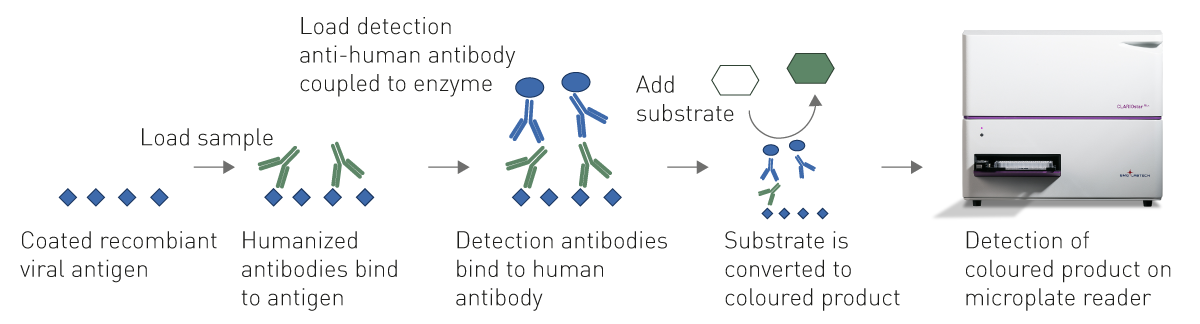
Contrary to classical ELISAs, homogenous immunoassays do not require washing steps to remove the unbound components from a well. This minimises both, handling steps and operation time, and makes them particularly suited for automation-supported screening. Homogeneous assays for virus detection include different TRF-, TR-FRET- and AlphaScreen-based assays as well as fluorescence polarization immunoassays.
The detection of viruses is a fundamental tool in diverse aspects of virology. Microplate readers provide a versatile platform, covering a multitude of assays used to analyse the molecular and cellular mechanisms of infection objectively and quantitatively. The range of virus detection methods or methods to analyze interactions and biological responses was even more broadened by recent developments in detection technology. Modern microplate readers further enhance these approaches, increasing sensitivity and speed, as well as providing the capability of real-time monitoring of virus infections in living cells for prolonged periods. And this is certainly not the end of the story: data quality and throughput will continue to benefit from further improvements in the alignment of microplate reader technology and microplate-based assays. In this way, the broad spectrum of existing assays could perhaps even be expanded to include previously unfeasible approaches.
Absorbance plate reader with cuvette port
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Upgradeable single and multi-mode microplate reader series
Flexible microplate reader with simplified workflows
Gene reporter assays are sensitive and specific tools to study the regulation of gene expression. Learn about the different options available, their uses, and the benefits of running these types of assays on microplate readers.
Do you want to study viral infectivity and screen for anti-virals? This blog post highlights the advantages of TCID50 assays for the determination of viral titers in microplate format.
This blog highlights some alternative viral detection methods to PCR. Find out, which can be run on microplate readers and what advantages the readout in microplates offers.
The dangerous viral outbreaks of recent years, such as SARS-CoV-2, illustrate the intensive research that virology requires. Find out more about available virus assays here.
Vaccines are powerful tools to fight viral diseases. Read more about the science behind vaccines and what we can expect from vaccine development in the future.