SPECTROstar Nano
Absorbance plate reader with cuvette port

Next generation sequencing (NGS) technologies enable genomes or parts of genomes to be sequenced in a matter of days or hours at great depth and low cost.
Not too long-ago progress in next-generation sequencing was defined as the quest for the $1000 genome.1 Today costs have plummeted even further, and several companies claim to have technologies that deliver $100 genomes and beyond.
These numbers reflect impressive developments in DNA sequencing technologies but in most cases represent the immediate costs of consumables on the sequencing instrument.2
DNA sequencing is also reliant on sample processing and DNA library preparation before sequencing as well as bioinformatics and data analysis after sequencing.
In this blog, we examine how quality control of nucleic acids on microplates before sequencing can facilitate next-generation sequencing and look at some of the emerging next-generation sequencing solutions available to researchers.
In essence, there are two main types of next-generation sequencing: short-read sequencing and long-read sequencing. Short-read sequencing is the most encountered type of next-generation sequencing where a genome is broken into many small fragments (typically 50 to 300 bases) before being sequenced. In contrast, long-read sequencing analyzes longer strands of DNA or RNA in one go without breaking the long strands into smaller fragments.
Short-read sequencing is a powerful tool for a wide range of applications and offers speed, cost-effectiveness and accurate base calling. This type of approach is particularly well suited to genomics applications like the analysis of single nucleotide polymorphisms (SNPs), gene expression and small-scale genetic variations. Long-read sequencing helps to resolve more structurally complex regions of sequence by its capability to sequence longer continuous strands of DNA. Long-read technologies can span repetitive or complex genomic regions that short-read sequencing may struggle to assemble accurately. This type of research is better suited to de novo genome assembly and provides longer contiguous sequences that reduce the need for extensive computational scaffolding and closing of gaps. This lends itself more to the generation of in-depth data about genome structure and function.
Here we will take a closer look at some of the pre-sequencing workflow where microplate readers can help to assure the quality control of nucleic acids prior to next generation sequencing. A typical workflow for genome sequencing is shown in Fig. 1. Quality control of nucleic acids is typically needed at the steps of DNA extraction and library preparation which we will consider in more detail.
Before sequencing can start, nucleic acid samples (DNA or RNA) must be prepared for analysis which involves their extraction from a biological sample. Nucleic acid samples need to be isolated and purified.
Extraction protocols are typically optimized for the type of sample. Nucleic acid extraction procedures include the use of phenol-chloroform, silica and magnetic bead-based extractions. Researchers will typically use commercially available kits for nucleic acid extraction compatible with their sample. There are no standard operating procedures for genomic DNA extraction and many options are available to choose from. Only a few studies compare the merits of one approach versus another.3 In the end, quality control of the extracted nucleic acid is the best indicator for integrity of the sample extraction method and the likelihood of success in the next steps. If RNA is being sequenced it is first converted to cDNA by reverse transcription.
The nucleic acid sample to be processed after extraction should be highly pure, with an A260/A280 ratio typically between 1.8 and 2.0 and should be as intact as possible. This allows for optimal fragmentation in subsequent steps.
Library preparation is an essential part of the workflow for next generation sequencing (Fig. 1). Sequencing libraries are typically made by generating fragmented DNA and ligating specialized adapters to both ends of the fragments. The adapters used will depend on the sequencing technology. Fragments can then be amplified and purified. In some cases, multiple libraries are pooled together and sequenced in the same run to save on resources.
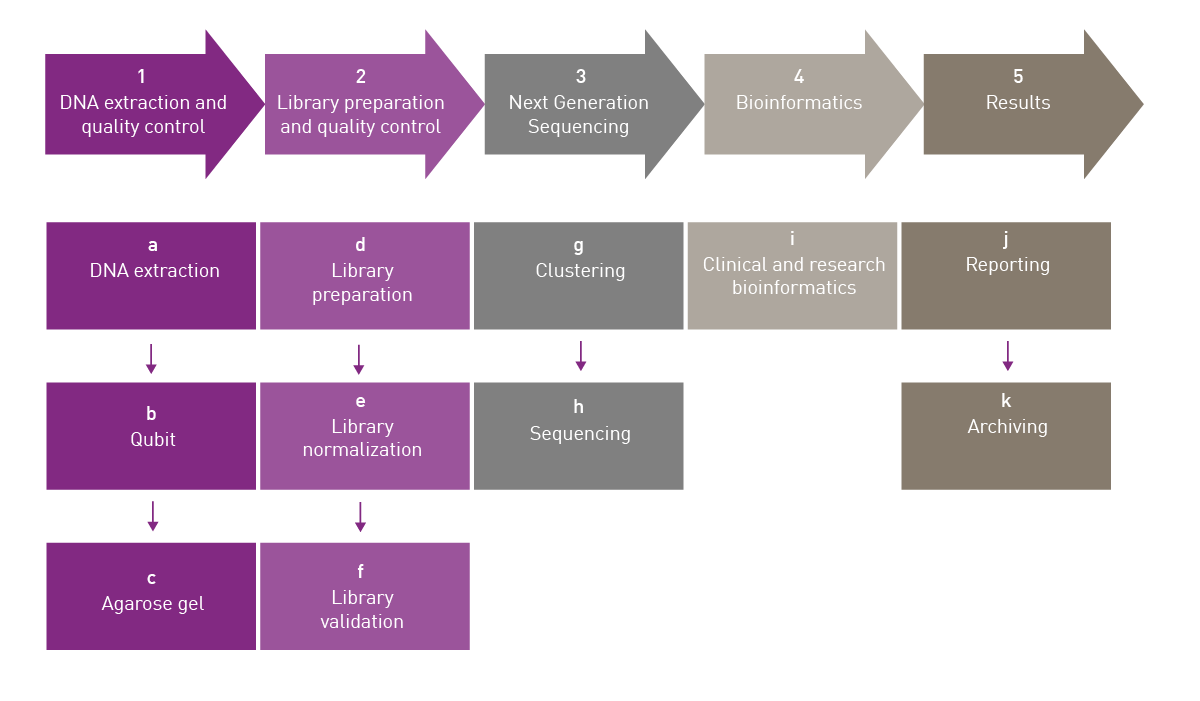
Library preparation and quality control comprises normalization and validation steps (Fig. 1). Library normalization is the process of diluting libraries of variable concentration to the same concentration before volumetric pooling. The goal is to ensure an even read distribution for all samples. For normalization, you first must determine the size of the library and quantify the individual libraries. The necessary dilutions are calculated and the normalized libraries pooled. Some library preparation kits offer a bead-based approach to normalization where manual normalization is not required.
In the validation step, the concentration of the library is determined by measuring the absorbance at 260 nm. The yield from a typical protocol should be between 500 and 1000 ng of DNA. The purity as measured by the A260/A280 ratio should be approximately 1.8. The overall preparation of the DNA library is essential for the success of the subsequent sequencing effort.
In next generation sequencing, it’s important to make sure all steps in the process are not only cost effective but also of the highest possible quality. Quality control of DNA extraction and DNA library preparation are therefore as essential as the fidelity of the DNA sequencing technologies since each represents a necessary step in the overall process.
Microplate readers are often used for DNA quality control before next generation sequencing is carried out. They provide a way to assess DNA concentrations and purity quickly and efficiently in high-throughput formats.
Nucleic acids can be quantified either by absorbance or fluorescence measurements. In practice, DNA samples are often measured as small volumes in microplate wells. For absorbance measurements, the microplate reader uses UV-visible spectrophotometry to measure the DNA at specific wavelengths typically around 260 nm. The DNA concentration in each well is calculated from the absorbance.
Microplate readers can also provide information about DNA purity. Absorbance measurements at different wavelengths are obtained: 260 nm (DNA); 280 nm (protein); 230 nm (contaminants, e.g. phenols) and ratios like A260/A280 and A260/A230 are calculated to indicate the presence of protein or contaminants that could affect downstream processes.
Acquisition of the whole UV-absorbance spectrum allows for DNA quantification and identification of contaminants like phenol (230 nm) as well as protein residuals (280 nm) to be carried out in one step. Measurements like this for DNA purity are typically performed on a microvolume spectrophotometer which requires only a microliter of sample. This type of detection device can only measure one sample at a time and is not compatible with high throughput.
As the only company dedicated exclusively to the manufacture of microplate readers, BMG LABTECH equips its instruments with a UV/vis spectrometer with spectral acquisition from 220 - 1000 nm. This enables the simultaneous acquisition of OD values at 230, 260 and 280 in less than 1 sec/well, much faster than filter- or monochromator-based devices. 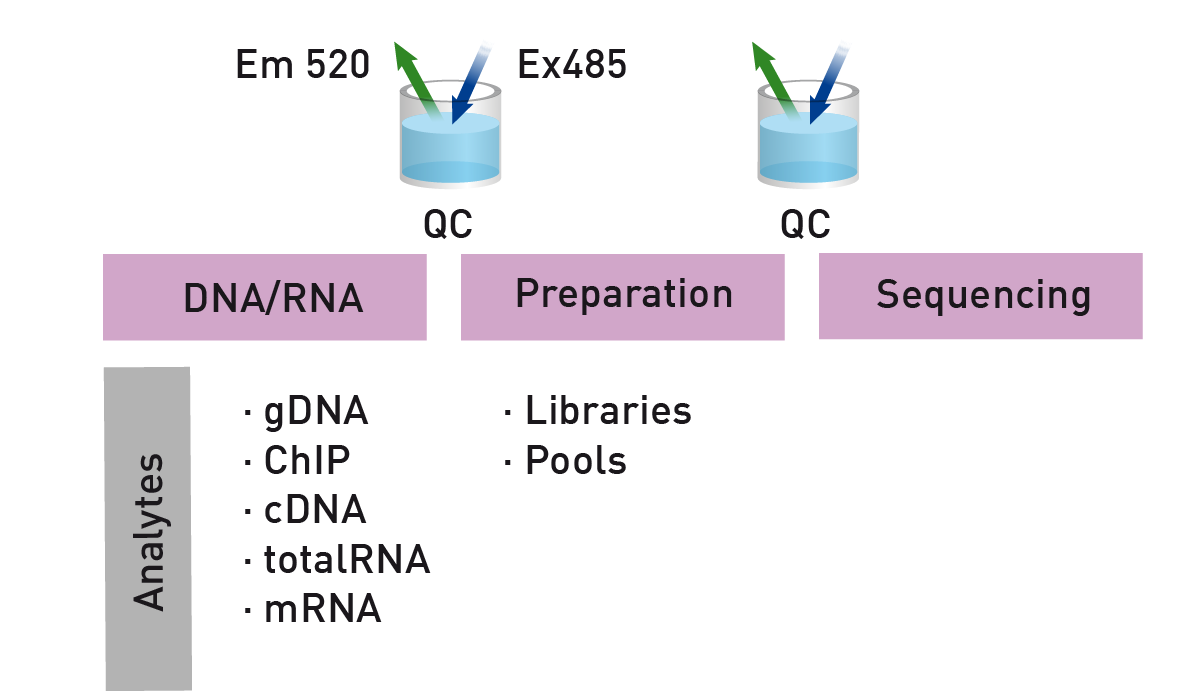
In most cases, DNA quantification using fluorescence is recommended for next-generation sequencing library preparation since it is more sensitive and more specific compared to UV absorbance quantification.
Standard methods use a dye that becomes highly fluorescent when associated with nucleic acids (Fig. 3). The gold-standard for nucleic acid quantification for next-generation sequencing purposes is Qubit™ which can be used to quantitate dsDNA and other nucleic acids.
Qubit assays are available for double- and single-stranded DNA, RNA, microRNA, crRNAs, and protein. Qubit DNA and RNA assays are also available with different quantification ranges. Different detection ranges of different Qubit kits allow accurate measurement across a wide range of concentrations. Find out more in our application note “DNA quantification using absorbance (A260) and fluorescent methods (Qubit™ and Quant-iT™/PicoGreen™).”
In a standard fluorometer, the Qubit dye is mixed with a nucleic-acid-containing sample and fluorescence is measured directly in the sample preparation tube. Typical fluorometers are limited to a single tube and detection is therefore at low throughput. However, both absorbance and fluorescence nucleic acid quantification methods can be transferred to higher throughput by using a microplate reader and 96-, 384- or 1536-well plates. 
The number of options for researchers looking for sequencing instruments has increased significantly in recent years and represents a rapidly changing and increasingly competitive market.4 The current landscape includes Illumina, Oxford Nanopore, PacBio, Singular Genomics, Complete Genomics (BGI), Element Biosciences, Ultima Genomics and other emerging and established companies.
Illumina’s sequencers are powered by its proven sequencing-by-synthesis (SBS) technologies and still have a significant part of the market but face increasing competition. SBS entails sequencing DNA by the synthesis of a complementary strand where each incorporated base is detected as it is added to the growing strand. SBS is used in Illumina’s MiSeq, HiSeq and NovaSeq systems which offer different options in scale and performance for different sequencing tasks. Illumina leads with benchtop machines such as the MiSeq, which is a fast, personal benchtop sequencer, with rapid run times and outputs intended for targeted sequencing and the sequencing of smaller genomes. The MiSeq was launched in 2011 and is the oldest next-generation sequencing sequencer on the market.
Oxford Nanopore Technologies provides real-time single molecule sequencing capabilities for DNA and RNA sequencing. The core technology involves passing a strand of DNA through a tiny pore while measuring changes in electrical current that occur as individual bases in a single strand pass through the nanochannel. These unique changes in current are used to identify the sequence of DNA and RNA. Oxford Nanopore’s most well-known platform is the MiniION which is complemented by the PromethION and GridION platforms to provide both short and long read sequencing options.
Over the years, PacBio has arguably been the leader in offering solutions for long-read sequencing with high accuracy (99.9%) comparable to the accuracy of short read technologies. Its HiFi powered technologies are driven by sequencing-by-synthesis methods for single molecule, real time sequencing. PacBio have complemented their strengths in long read sequencing by the recent acquisition and launch of a short-read sequencing-by-binding technology on the Onso platform.
Singular Genomics launched the G4 sequencing platform at the end of 2021. G4 is a sequencing-by-synthesis platform which offers high data output for short reads in a benchtop instrument.
Complete Genomics has developed and launched a sequencing-by-synthesis and ligation method that combines rolling circle amplification with DNA Nanoball technology (home to the ligation steps). The combination of these two steps allows for longer reads at high accuracy. The company offers instruments at all scales from the benchtop to its T20 platform for ultra-high throughput sequencing.
Element Biosciences was founded in 2017 and launched the AVITITM system in 2022. AVITITM is a cost-effective benchtop sequencing-by-synthesis technology that offers short reads and smaller long reads up to approximately 10 kb.
Ultima Genomics has developed a sequencing-by-synthesis technology that produces single read data off a flow-based circular disc. The sequencing platform follows the incorporation of one base at a time and does not require blocking of synthesized sequences by reversible terminator deoxyribonucleoside triphosphate molecules.
The next-generation sequencing landscape of technologies changes very quickly and any description of the competitive landscape can be quickly out of date.
Next-generation sequencing technologies have evolved considerably since the initial completion of the Human Genome Project in 2003. In 2022, the hard-to-sequence telomere regions were finally sequenced, and one could argue that this represents a next-generation human reference genome sequence put together with the help of long read sequencing from PacBio.
Enhancements in chemistry, cost, throughput and availability are driving the rise of new, varied technologies to address existing applications and those that were not formerly possible. These include but are not limited to:
Developments in these areas support many applications in clinical diagnostics, cancer genomics, evolutionary biology, agriculture, personal genomics, pharmacogenomics, environmental science, and more.
In the RNA world, next-generation sequencing supports a wide range of applications that span gene expression and which include gene expression profiling, differential gene expression analysis, alternative splicing, the discovery of non-coding RNAs, single-cell RNA sequencing, and functional genomics. These applications find uses in cancer research, the study of infectious diseases, developmental biology, neuroscience, environmental biology, agriculture, drug discovery, SNP and more.
It is undeniable that the costs of next-generation sequencing alone have plummeted at a remarkable rate. Although huge strides are being made, limitations remain for many next-generation sequencing technologies, particularly for sequencing long homopolymer stretches and GC-rich regions where coverage and accuracy across the genome still have issues. From a technological perspective, the time required to sequence and analyse data still limits the use of next-generation sequencing in clinical applications in which time is an important factor. Costs beyond next-generation sequencing alone can also increase.
What is the preferred BMG LABTECH microplate reader for specific needs and applications related to the quality control of DNA or RNA for next-generation sequencing? BMG LABTECH offers a range of detection devices for sensitive absorbance and fluorescence measurements suitable for quality control of nucleic acids. They combine speed and the acquisition of complete absorbance spectra making them ideal for nucleic acid quantification.
All BMG LABTECH microplate readers have exceptionally fast reading capabilities. Collectively, these multi-mode readers combine high performance with miniaturized assays, short measurement times, and offer considerable savings on materials and other resources.
For more information on how BMG LABTECH microplate readers can help increase throughput and sensitivity in nucleic acid quantification assays, have a look at our brochure Accelerating your nucleic acid quantification for NGS.
Therefore, these types of detection devices can be used to perform estimations of concentration and quality using absorbance as well as specific measurements of DNA by fluorescence.
Absorbance plate reader with cuvette port
Powerful and most sensitive HTS plate reader
Most flexible Plate Reader for Assay Development
Upgradeable single and multi-mode microplate reader series
Flexible microplate reader with simplified workflows
Life in the depths of the ocean operates under extreme conditions. Find out how proteins from deep-sea luminescent organisms are useful for measurements on microplate readers.
Measuring light scattering is an accurate and expedient way to determine drug solubility. Find out how the NEPHELOstar® Plus can be used for early drug solubility screening at high throughput.
Mitochondrial toxicity can have devastating effects on the cell and life. Find out how microplate readers can be used to assess mitochondrial health and how this impacts disease and drug discovery.
Find out how microplate readers can be used to measure histone deacetylase (HDAC) activity and assist drug discovery.
Read how various nucleic acid quantification methods, ranging from UV absorbance and fluorescent dyes to amplification methods, can be performed on BMG LABTECH microplate readers.