Introduction
Various biological processes depend on the interaction of proteins with nucleic acids. These comprise packaging of DNA, transcriptional regulation, DNA replication and DNA repair. The binding of proteins to nucleic acids are hitherto detected by binding assays such as electrophoretic mobility shift assays (EMSAs), fluorescence polarization (FP) assays or chromatin immunoprecipitations (ChIP). However, these methods are either laborious (EMSA, ChIP) or limited to specific targets (EMSA, FP, ChIP). A novel fluorescence binding assay detects interactions of nucleic acids with proteins to identify binding, sequence and structure specificities or dissociation constants. The microplate assay is a quick and cost-effective alternative and can be read on a fluorescence reader with the capability to scan wells such as the FLUOstar® Omega.
Assay Principle
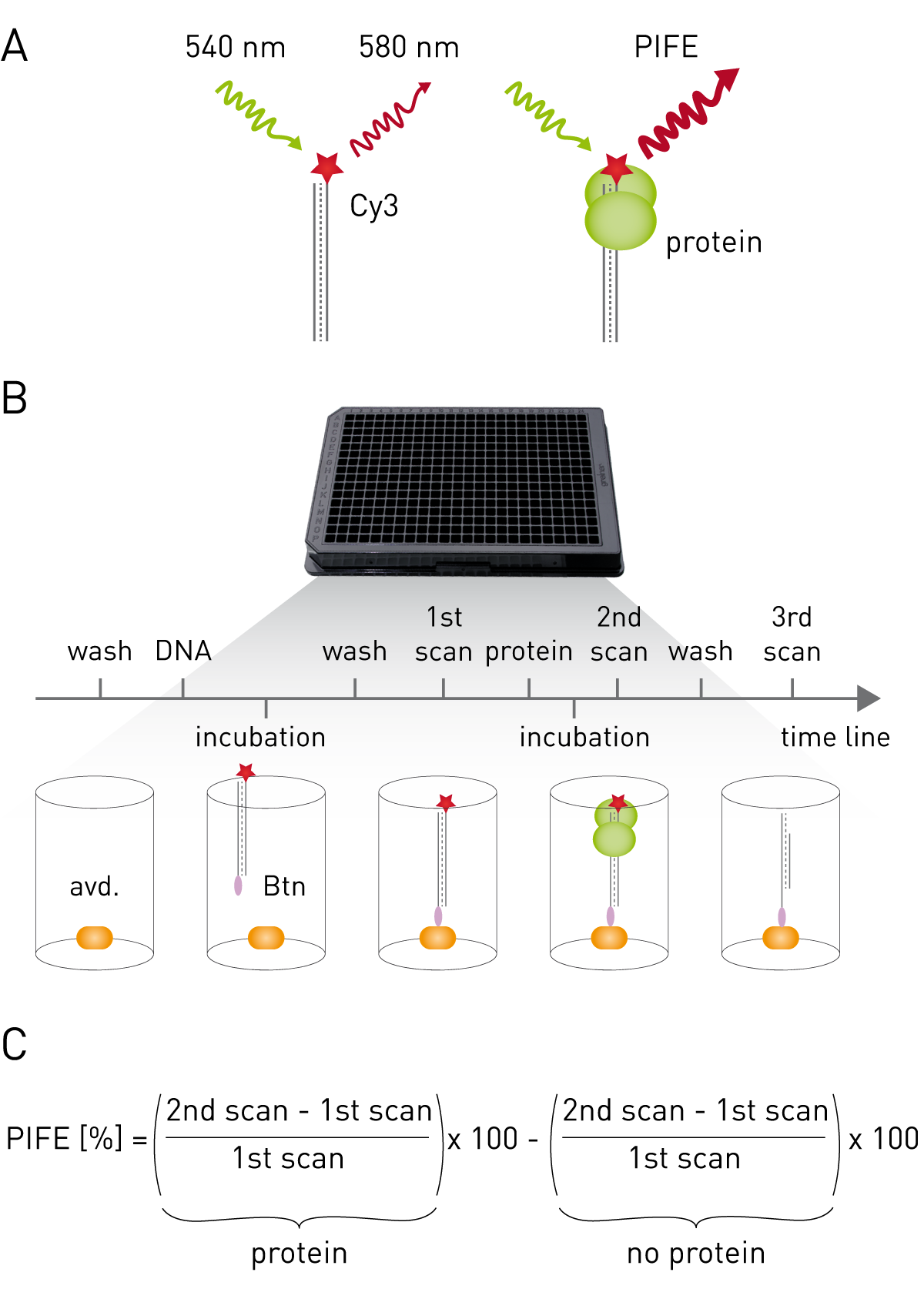
Cyanine dyes appear as non-fluorigenic cis-isomers and as fluorescent trans-isomers dependent on their local environment1. The microwell protein-induced fluorescence enhancement (mwPIFE) assay exploits this phenomenon as it uses Cy3-bound oligomers immobilized onto the bottom of a microplate. Protein-binding in proximity to Cy-3 sterically hinders formation of the non-fluorescent cis-isomer leading to increased fluorescence (Fig. 1A). Workflow and calculation of PIFE value are shown in Figure 1B and C.
Materials & Methods
- Black 96 well microplate (Greiner, FluotracTM 600, high binding, #655077) coated with 100 µl NeutrAvidin (Pierce, binding capacity ~15pmol D-biotin/well) and blocked with SuperBlock blocking buffer (ThermoScientific, #15117)
- Proteins*: BamHI (ThermoScientifi c), XPF/ERCC1 (expressed in Escherichia coli strain BL21 (DE3)), KU70/KU80 (expressed in HI5 insect cells)
- Cy3-labeled oligonucleotides (IDT)*
- FLUOstar Omega microplate reader (BMG LABTECH)
The Cy3-labeled oligonucleotides (100 μl, 25nM) were incubated on the plate for 2 h to bind and sub-sequently washed three times to remove residual nucleic acids. A first scan determined baseline fluorescence, a second scan detected binding of the protein to the oligonucleotide after incubation with 100 μl of the protein solution.
Instrument settings
| FLUOstar Omega | ||
|
Optic settings
|
Fluorescence Intensity, well scan mode, top optic | |
| Excitation: | Ex540-10 | |
| Emission: | Em580-10 | |
| Gain: | 2000 | |
|
General settings
|
Settling time: | 0.1 s |
| Number of flashes per scan point: |
10 | |
|
Scan settings
|
Matrix | |
| Scan points: |
10 x 10
|
|
| Diameter: |
6 mm
|
|
Results & Discussion
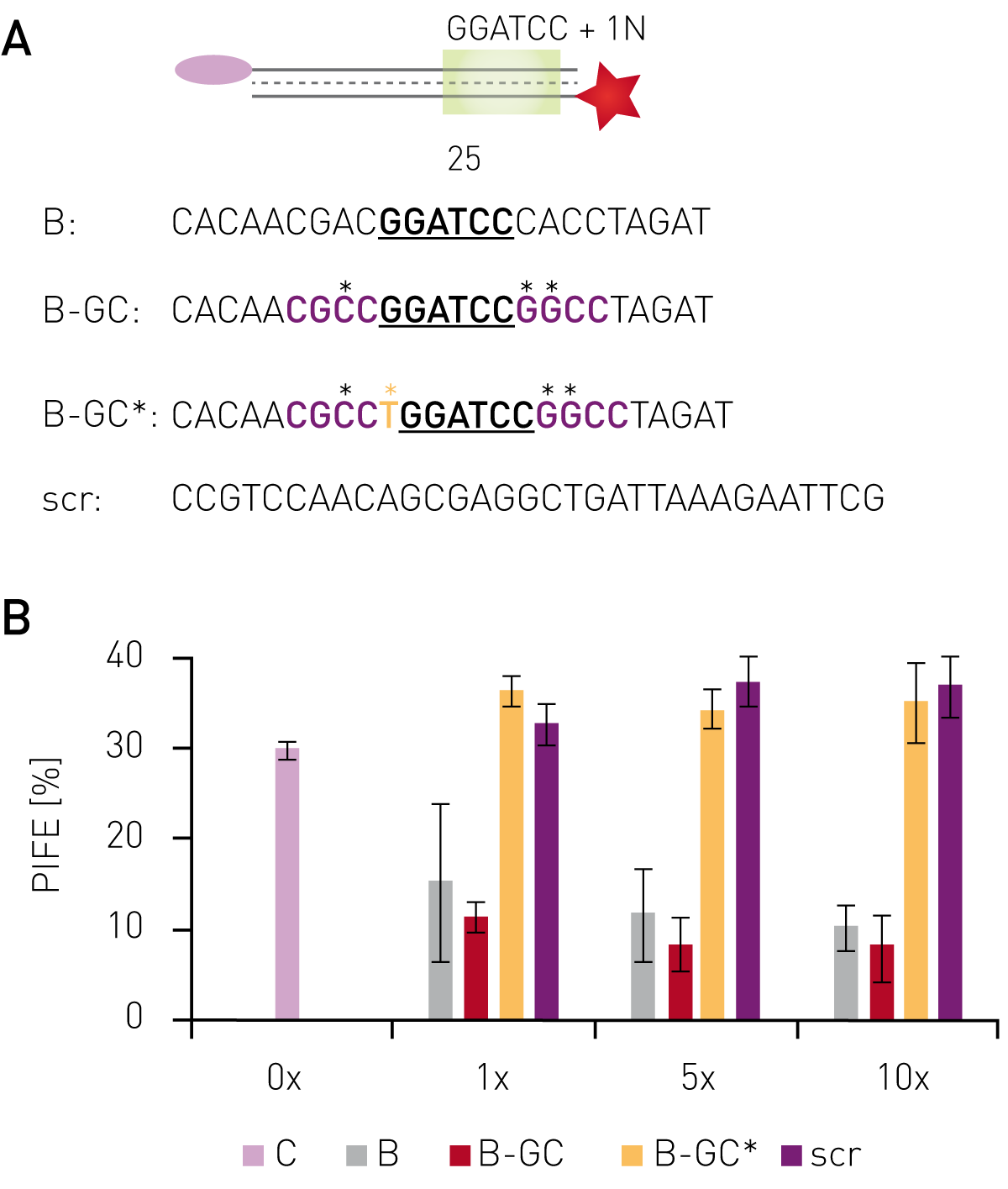
Using protein-induced fluorescence enhancement in microplates, the binding of the endonuclease BamHI to its recognition site was verified by a PIFE-value of 30 % (Fig. 2B). Addition of non-labelled oligonucleotides containing the recognition site, but sequence changes outside the recognition motif decreased the PIFE value down to 5-15 %, depending on concentration and sequence. This indicates lower BamHI-binding to the Cy3-labelled oligo due to competitive binding to unlabeled oligos. Non-labeled probes bearing mutations inside the recognition site gave a PIFE-value of >30 %, representing unperturbed binding of BamHI to the wild-type recognition site.
Having shown that mwPIFE detects sequence pre-ferences of protein-DNA binding, preferential binding of proteins to specific DNA structures was investigated. The XPF/ERCC1 heterodimer incises damaged DNA for repair and specifically binds structures of damaged DNA. A truncated XPF/ERCC1 heterodimer retaining the DNA-binding site was used to determine structure specific interaction. The dimer minimally bound ssDNA or short, blunt ended DNA if competing with the probe displayed in Figure 3B. Reduction of the PIFE-value due to binding of the competing DNA-structure was observed for 10 nt 3’overhang, a hairpin and a splayed DNA-structures (Fig. 3).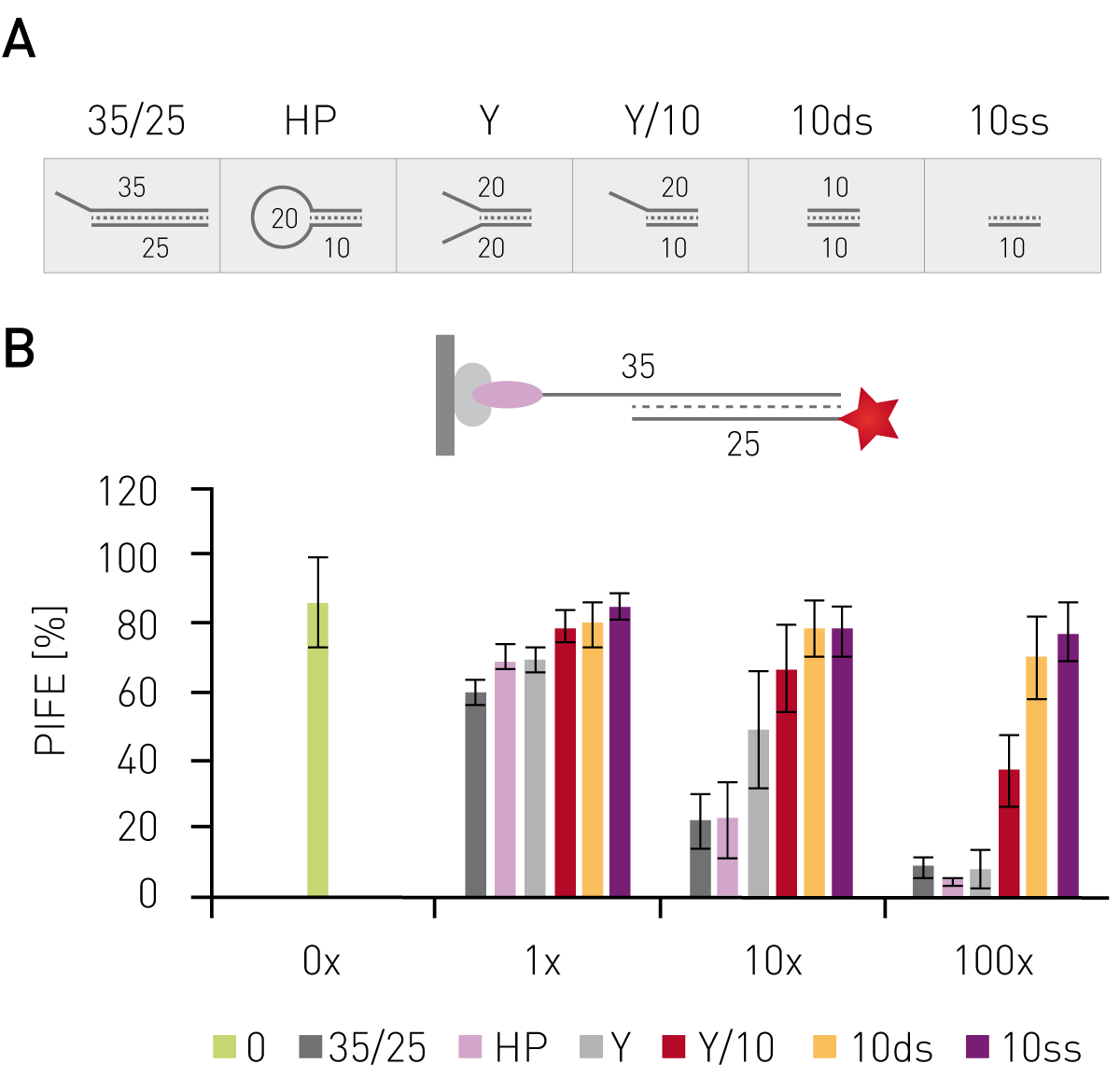 The KU70/KU80 heterodimer is responsible for binding broken dsDNA ends, thereby protecting them from degradation and keeping both ends in proximity to allow for ligation. Increasing the concentration of KU-heterodimer resulted in higher PIFE-values and allowed for the determination of the equilibrium constant KD (Fig. 4).
The KU70/KU80 heterodimer is responsible for binding broken dsDNA ends, thereby protecting them from degradation and keeping both ends in proximity to allow for ligation. Increasing the concentration of KU-heterodimer resulted in higher PIFE-values and allowed for the determination of the equilibrium constant KD (Fig. 4).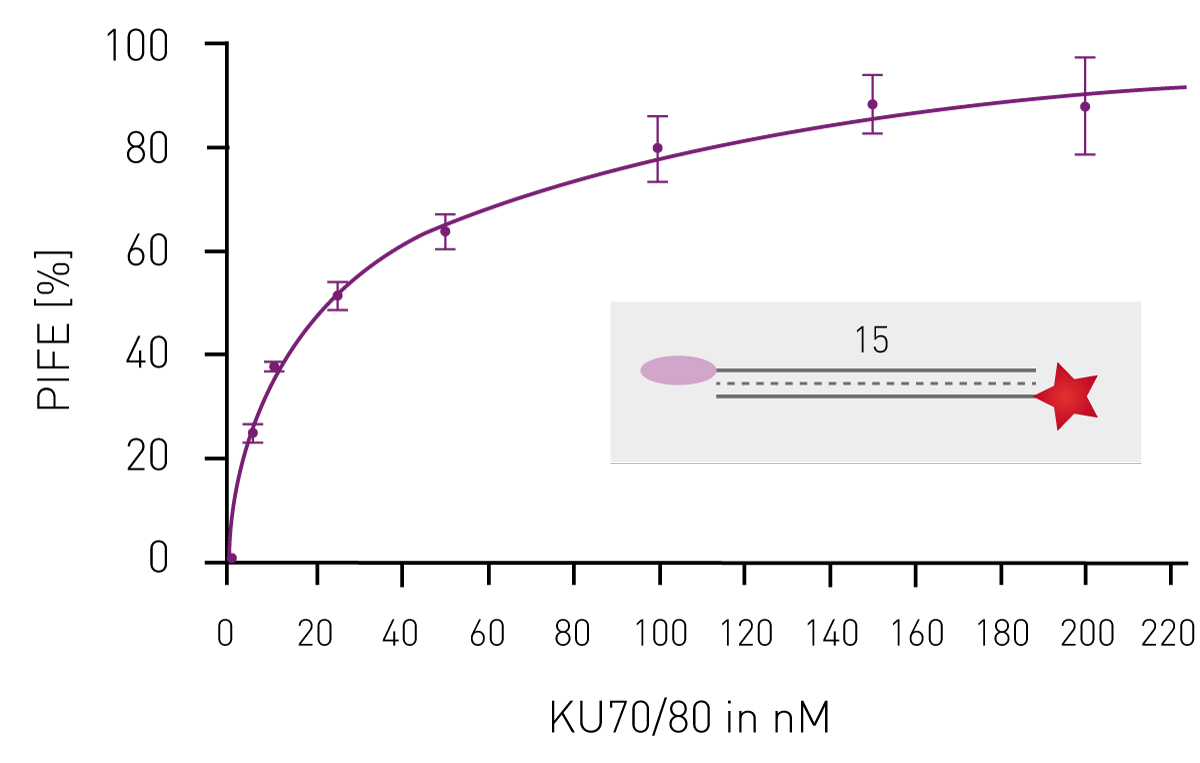 Conclusion
Conclusion
The mwPIFE detects interaction of nucleic acids with proteins and reports on sequence and structure specificities as well as on binding constants. The change in fluorescence intensity needs to be captured on a huge surface of the well to provide stable measurements. This has been enabled by the well-scan function of the FLUOstar Omega combined with its high sensitivity. The simple and cost-effective assay broadens the application to detect structure-specific bindings. Its plate format accelerates the determination of protein-nucleic acid interactions.*
References
- Hwang H. (2014) Chem Soc Rev. 43(4): 1221–1229
- Valuchova S. (2016) Sci Rep. 23;6:39653.
*For further information and when publishing data using the mwPIFE please refer to Valuchova et al.2