Introduction
A carefully maintained intracellular redox equilibrium is necessary for the proper function of a living cell. Oxidative stress, or a disturbed intracellular redox balance, has been linked to several diseases, including neurodegenerative diseases such as Alzheimer’s disease, rheumatoid arthritis, acute cerebrovascular disorders, diabetes and cancer, and accumulates in normal ageing. In oxidative stress there is increased production of oxidising species, primarily reactive oxygen species (ROS), which outperforms the ability of the intrinsic antioxidant defence mechanisms of the cell to neutralise them. Neurons are highly susceptible to oxidative damage due to high levels of ROS production through respiration and metabolism, presence of lipid peroxidation-susceptible polyunsaturated fatty acids in high concentrations and relatively low levels of some antioxidant enzymes, like catalase.
The main sulphur-centred antioxidant systems are the glutathione (GSH) and the thioredoxin (Trx)- reduction pathways. With both pathways, oxidative stressors such as hydrogen peroxide are reduced by NADPH, via a series of oxidations and reductions involving glutathione (GSH), Glutathione peroxidase (GPx) and Glutathione reductase (GR) for the former, and Peroxiredoxins (Prx) – or other proteins-, Thioredoxin (Trx) and Thioredoxin reductase (TrxR) for the latter. The GSH pathway is more effective in reducing small disulfide molecules and direct interactions with ROS, whereas Trx is more effective in repairing oxidised proteins by reducing their exposed disulfides while its own active site cysteine residues get oxidised (and reduced by TrxR).
The thioredoxin system protects cells against H2O2-induced cell death and its inhibition promotes oxidative stress, while thioredoxin-overexpressing mice display less oxidative brain damage following ischemia and live longer.
We are interested in thioredoxin activity in neurons and its potential changes under different conditions. However, because primary cortical cultures that we use as a model are mixed cultures of neurons and glial cells, we wanted to establish what the level of thioredoxin activity is in either cell type (neurons versus glia).
Materials & Methods
All materials were obtained through normal distribution channels from the manufacturers stated.
- Transparent 96-well plates (Greiner)
- BMG LABTECH microplate reader
- Leica DMI6000B inverted epifluorescence microscope
Neuronal cultures and trophic deprivation
Cortical rat neurons were cultured as described from E21 rats. Glial cells were cultured by plating rat cortical homogenate at half the density of that for neuronal cultures and grown in DMEM (Dulbecco’s Modified Eagle’s Medium) including 10% Fetal Calf Serum (Invitrogen) and antibiotics (Penicillin/Streptomycin 1:100, Sigma). No AraC (Cytosine β-D-arabino-furanoside hydrochloride, a DNA synthesis inhibitor, Sigma) was added to glial cultures in contrast to neuronal ones at DIV4 (Day In Vitro 4). Neurons were killed in glial cultures by overnight incubation in 1 mM NMDA (N-methyl-D-Aspartic acid, Calbiochem) in minimal medium (TMo) on DIV7. Trophic deprivation was done in both types of cultures after a culturing period of 8-10 days during which cortical neurons develop a network of processes, express functional NMDA-type and AMPA/kainate-type glutamate receptors, and form synaptic contacts. Cells were transferred into defined medium lacking trophic support, hereafter “TMo”: 10% MEM (Invitrogen), 90% Salt-Glucose-Glycine (SGG) medium (SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM Glycine, 30 mM Glucose, 0.5 mM sodium pyruvate, 0.1 % Phenol Red; osmolarity 325mosm/l).
Thioredoxin Activity Assay
The thioredoxin “insulin-reducing assay” was performed with some modifications. Briefly, cells were grown for 24 h in minimal medium TMo, then lysed by scraping on ice in 150 μl lysis buffer/35 mm dish (20 mM HEPES pH 7.9, 100 mM KCl, 300 mM NaCl, 10 mM EDTA, 0.1 % Triton X-100, 1:100 Protease Inhibitor Cocktail III- Calbiochem) after being washed twice in pre-warmed TMo medium. Cell lysates were collected in microcentrifuge tubes and cell debris discarded by centrifugation for 3 min at 10000 rpm at 4 °C. The appropriate volume of lysate containing 30 μg protein was diluted in lysis buffer to a final volume of 34 μl. [We tested a series of protein concentrations to select one that fell within the linear range of the reaction, starting with amounts of protein that had been shown to be in the linear range for this assay previously. Alternatively or in addition, a standard curve using purified thioredoxin can be included to confirm the linear range of the assay.] 1 μl DTT activation buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 1 mg/ml BSA, 2 mM DTT) was added to all tubes. After mixing, the tubes were incubated at 37°C in a waterbath for 15 min, for reduction of endogenous thioredoxin. 20 μl reaction mixture (200 μl 1M HEPES pH 7.6, 40 μl 0.2M EDTA, 40 μl NADPH 40 mg/ml and 500 μl insulin 10 mg/ml) was then added to each tube, as well as 0.5 units of Trx reductase or H2O for negative controls, and the samples were incubated at 37°C for 20 min. The reaction was stopped by addition of 250 μl Stop Buffer (6 M guanidine HCl, 1 mM DTNB (Sigma) in 0.2 M Tris-HCl pH 8.0), 200 μl of sample were transferred to a 96-well plate and absorbance was measured at 405 nm in a BMG LABTECH microplate reader.
Assay Principle
(1) Trx-S2 + DTTred → Trx-(SH)2 + DTTox
(2) Trx-(SH)2 + Insulin-S2 → Trx-S2 + Insulin-(SH)2 TrxR
(3) Trx-S2 + NADPH + H+ → Trx-(SH)2 + NADP+
(4) Insulin-(SH)2 + DNTB → Insulin-S2 + 2TNB(Bright yellow)
Endogenous Thioredoxin (Trx) in the samples is first reduced by dithiothreitol (DTT, red for reduced and ox for oxidised forms); (reaction 1). After the addition of NADPH, Insulin and Thioredoxin Reductase (TrxR), Trx reduces insulin disulfides (reaction 2). In the presence of TrxR and at the expense of NADPH, Trx can be reduced and brought back into the system (reaction 3). When the reaction is stopped the sulfhydryl groups are derivatized to TNB (reaction 4) which gives an intense yellow colour.
Immunocytochemistry and cell counting
Cell cultures were fixed with 3 % paraformaldehyde and 4 % sucrose for 20 minutes at room temperature (RT) and then permeabilized for 5 min in phosphate-buffered solution (PBS) supplemented with 0.5 % NP-40. Cells were incubated overnight at 4°C in PBS with primary antibodies against glial fibrillar acidic protein (GFAP) (1/2000; Sigma) and Neuronal Nuclei (NeuN; 1/15; Chemicon), to stain glial cells (astrocytes) and neurons, respectively. NeuN staining was completed with 1 h incubation with biotinylated anti-mouse (1/200); (Jackson ImmunoResearch) antibody, followed by 1 h incubation with Cy3-coupled streptavidin (1/500).
GFAP staining was completed with Alexa 488-coupled anti-rabbit (1/500) antibody (Jackson ImmunoResearch) incubated for 1 hour at RT. After mounting with DAPI-containing solution (Vectashield; Vector Laboratories), cells were observed and images were taken using the 10X or 20X objective of a Leica inverted epifluorescence microscope driven by LAS software. Cells were counted (blind) to establish the total number of cells per culture type (DAPI-stained) and the percentage of NeuN-positive and GFAP-positive cells, using ImageJ software.
Results & Discussion
We performed thioredoxin activity assays in parallel in pure glial cell cultures and mixed neuronal-glial cultures (thereafter referred to as “cortical”) that were under trophic deprivation for 24 h. Absorbance was measured in a BMG LABTECH microplate reader and a control reaction without Thioredoxin reductase enzyme was performed for each sample and this value was subtracted from the value of the sample containing TrxR.
At the same time, we used immunostaining to establish the number of cells present in each type of culture.
From the thioredoxin activity assay data combined with the cell counts from parallel dishes, the thioredoxin activity of a neuron in a mixed cortical culture, relative to the activity of a glial cell was calculated using the formula: “(number of glial cells/cort. dish)*Glial activity + (number of neuronal cells/cort.dish)*Neuronal activity = (fold-increase of Trx activity in cortical compared to glial culture)*(number of glial cells/glial dish)*Glial activity”. Thus, the thioredoxin activity of a neuron is 3.8 ± 0.86 fold higher than that of a glial cell (Figure 1). Since the glial cells in a mixed cortical culture represent 4-9 % of the total number of cells, the thioredoxin activity represented by glial cells is only 1-2.5 % of the total thioredoxin activity in that culture.
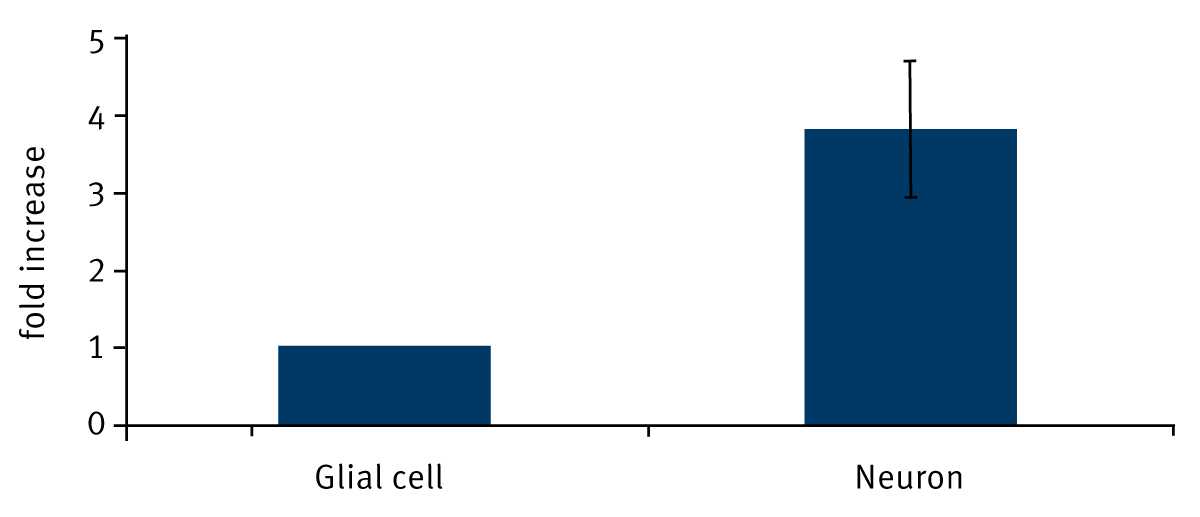
Conclusion
In our system only 1-2.5 % of the thioredoxin activity in primary cortical cultures is attributable to glial cells, even though glial cells represent 4-9 % of the total cell number. Therefore, neurons are responsible for the vast majority of thioredoxin activity in mixed rat cortical cultures. The thioredoxin activity assay can easily be performed on a BMG LABTECH microplate reader.