Introduction
Transglutaminases (TGs) form a family of enzymes that catalyze various posttranslational protein modifications such as crosslinking, esterification, and deamidation in a Ca2+-dependent manner. Their main function is the formation of covalent Nε-(g-glutamyl)lysine bonds within or between polypeptides to stabilize protein assemblies. The activity of these enzymes is crucial for tissue homeostasis and function in a number of organ systems, and the lack of or excessive crosslinking activity has been linked to human disease processes.
Here we perform kinetic measurements using recombinant TG2 and a fluorescent peptide model substrate in a format suitable for high-throughput analysis. This assay principle can be applied to kinetic studies on closely related enzymes including TG6 and can be optimised by modification of the backbone peptide sequence.
Assay principle
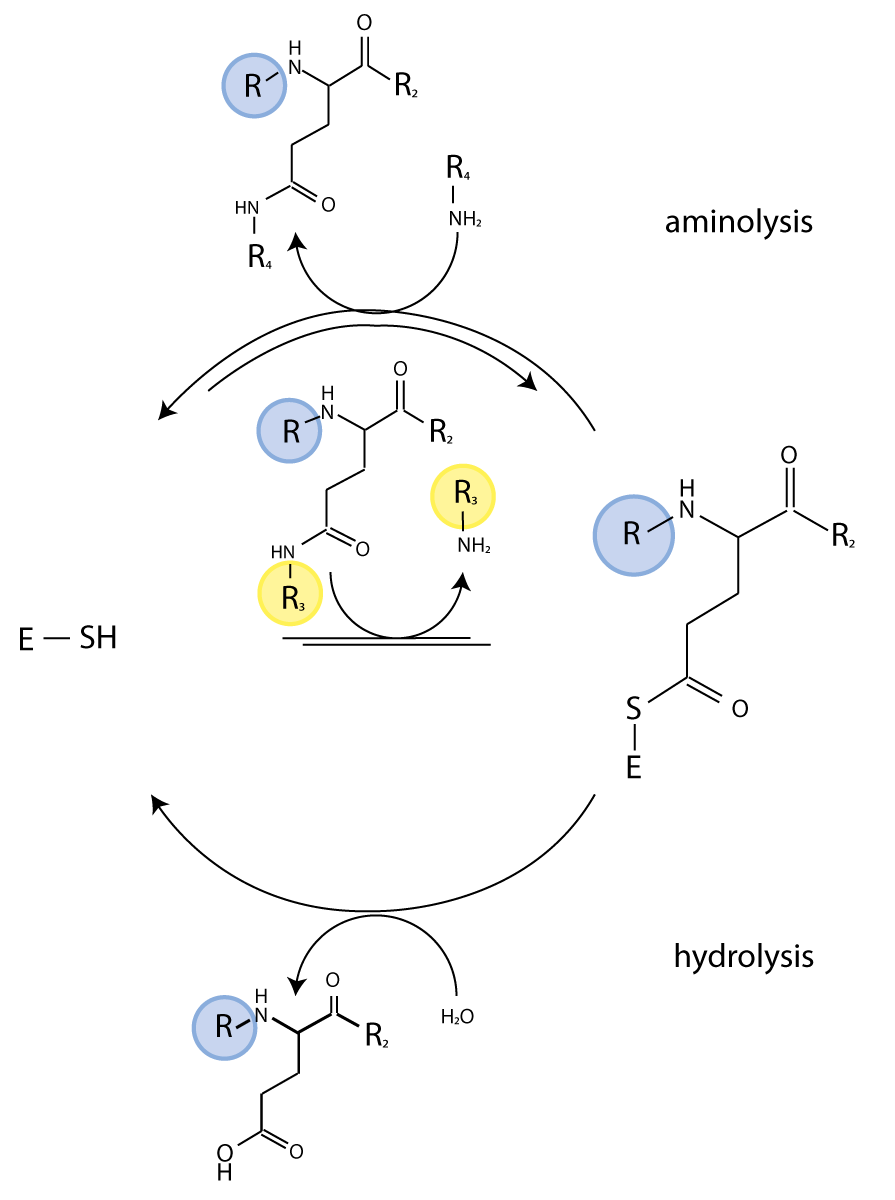
The TG enzymatic reaction is a two-step process. The thioester intermediate of the enzyme formed with the substrate in the first step subsequently reacts with a nucleophile to regenerate active enzyme and release a ‘crosslinked’ polypeptide. The second step is reversible and, in the presence of an excess of crosslinked substrate, TG catalyzes isopeptide bond hydrolysis. We have exploited this latter activity for real-time monitoring of TG activity and characterize the effect of potential regulators on TG activity.
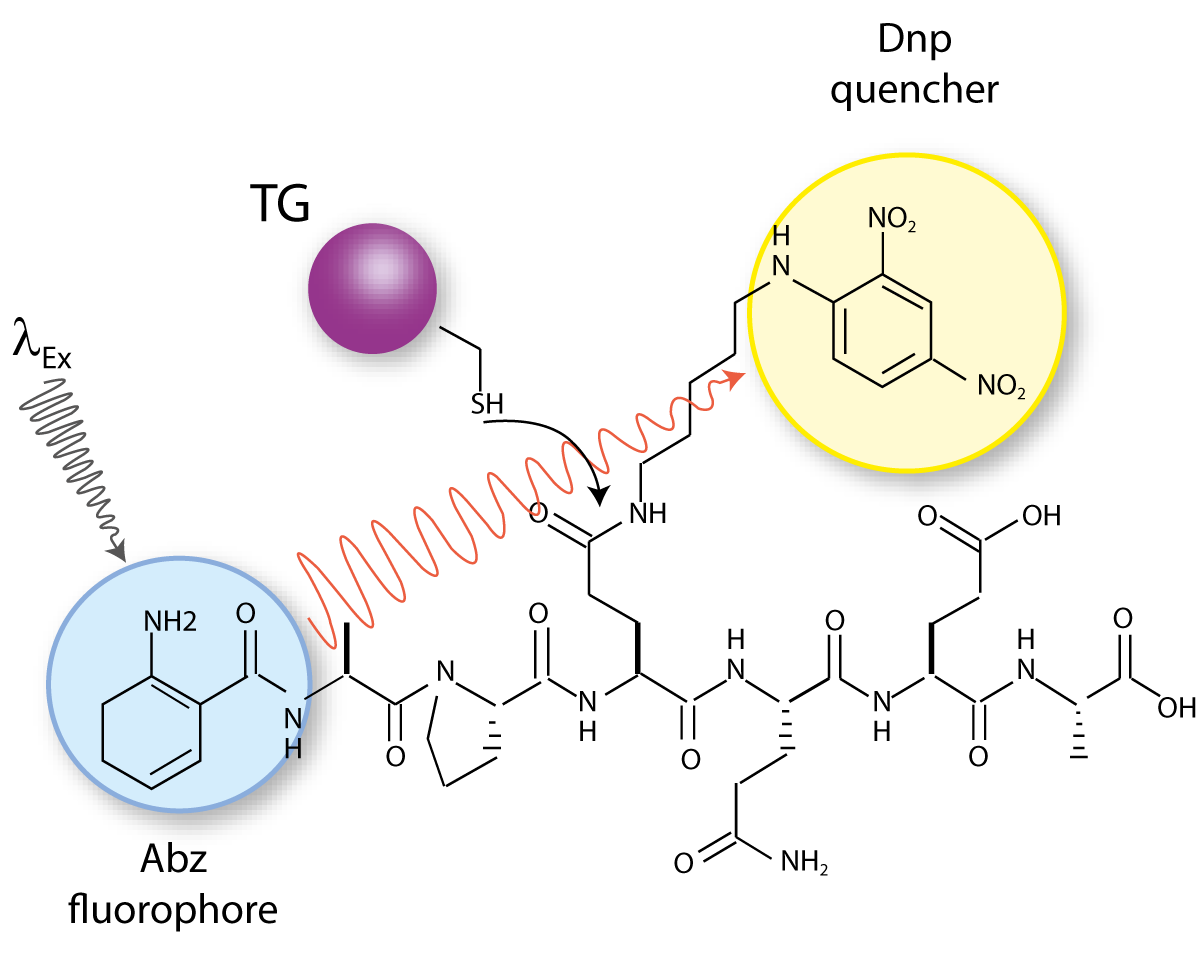
Abz-APE(g-cad-Dnp)QEA is a quenched fluorescent probe derived from a known glutamine donor substrate that mimics a crosslinked TG reaction product. In this peptide the fluorophore (2-aminobenzoyl (Abz)) is quenched by a 2,4-dinitrophenyl-cadaverine (cad-Dnp) substituent on the first Gln residue, essentially replacing the Lys side chain in Nε-(g-glutamyl)lysine linked peptides (Fig. 1). TG2-catalysed hydrolysis of the isopeptide bond releases the cad-Dnp moiety (Fig. 2) and consequently generates an increase in light emission at λmax=418 nm from the Abz group. The thioester enzyme intermediate formed is subsequently deacylated through either aminolysis or hydrolysis. Specificity of the reaction is guided by the amino acid residues that surround the reactive Gln residue.
Materials & Methods
- Microplate reader from BMG LABTECH
- Black optical bottom 96-well plates (Nunc)
- Abz-APE(g-cad-Dnp)QEA TG2 substrate (Zedira). 50 mM stock in DMSO
- Transglutaminase 2, 1 mg/ml stock, from Zedira
Assay buffer
The assay buffer consists of 62.5 mM Tris/HCl, pH 7.4, 125 mM NaCl. Add glycine methylester (or alternative amine donor substrate) and adjust pH immediately before use (at 37˚C). Include DTT to prevent oxidative inactivation of TG.
Test protocol
Prime the microplate reader injectors with 20 mM CaCl2 for enzyme activation (inj. 1) and H2O or 20 mM MgCl2 for control reaction (inj. 2). Pre-warm assay buffer and plate to 37˚C and equilibrate instrument chamber at 37˚C. Dilute substrate Abz-APE(g-cad-Dnp)QEA (1:800) in assay buffer and add 80 µl of mixture into wells of the 96-well plate. Add desired amount of enzyme, e.g. 1 mg of TG2, and make up volume with H2O to 90 µl. Transfer plate immediately into the reader and start program.
Reaction mixture (final concentrations)
The final assay volume is 100 µl and consists of 1- 100 µg/ml TG2, 50 µM Abz-APE(g-cad-Dnp)QEA, 10-55 mM glycine methylester or alternative nucleophile, as well as 1-5 mM DTT. After injection, there is 2 mM CaCl2 present in samples.
Instrument settings
|
Mode:
|
Fluorescence Intensity, plate mode
|
|
Filters:
|
Excitation: Ex320
Emission: 440-10
|
|
Optics:
|
Top
|
|
No. of flashes:
|
20
|
|
Cycles:
|
90
|
|
Cycle time:
|
40 s (for 12 wells)
|
|
Injection cycle:
|
10
|
|
Injection volume:
|
10 µL
|
|
Shaking:
|
5 s after each cycle
|
|
Temperature:
|
37°C
|
Results & Discussion
After Ca2+ injection an increase in fluorescence can be observed dependent on the concentration of enzyme in the sample (Fig. 3).
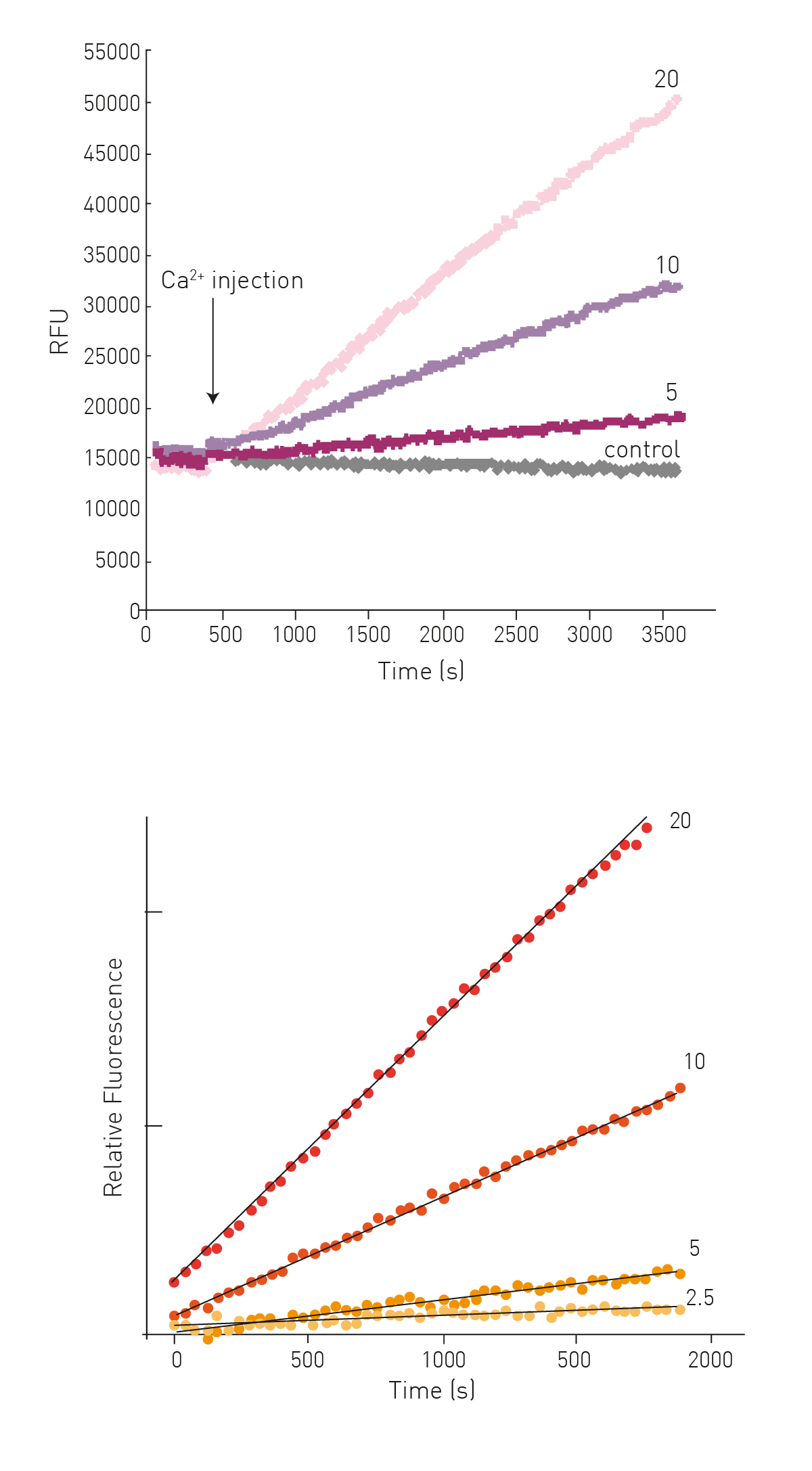
TG2-mediated substrate conversion is linear for >30 min and initial reaction rates can be derived from linear regression of the first 15-25 data points (Fig. 3).
Conclusion
Reported here are optimized experimental conditions for determination of TG2 isopeptidase activity with the fluorescent model substrate Abz-APE(gcad-Dnp) QEA using the BMG LABTECH microplate readers to produce an assay that is rapid, direct, and sensitive. Automated injection of Ca2+ for enzyme activation combined with the ability to continuously measure fluorescence intensity over a considerable time period with limited photo-bleaching facilitates the acquisition of kinetic data. Small sample size and plate format make the assay cost-effective and adaptable to high-throughput analysis.
Acknowledgements
This work was supported by grants from Coeliac UK and Arthritis Research UK (18461), and PhD studentships from Cardiff University/the President’s Scholarship scheme to MA and AH.
Check an additional customer testimonal on transglutaminase studies in celiac disease research.