Introduction
Human Lys-specific Histone Demethylase 1 (LSD1) promotes the demethylation of mono- or di-methyl-Lys4 on histone H31: as chromatin remodelling enzyme, LSD1 is found in complex with several partners, one of which being the REST Co-repressor 1 (CoREST1). We used the fluorescence polarization (FP) as a bio-physical tool to analyse the binding properties of LSD1-CoREST1 (LC1) hetero-dimer to H3-derived peptides (Fig. 1), that have been conjugated at the C-terminal with the fluorescent label TAMRA (5-Carboxytetramethylrhodamine).
Assay Principle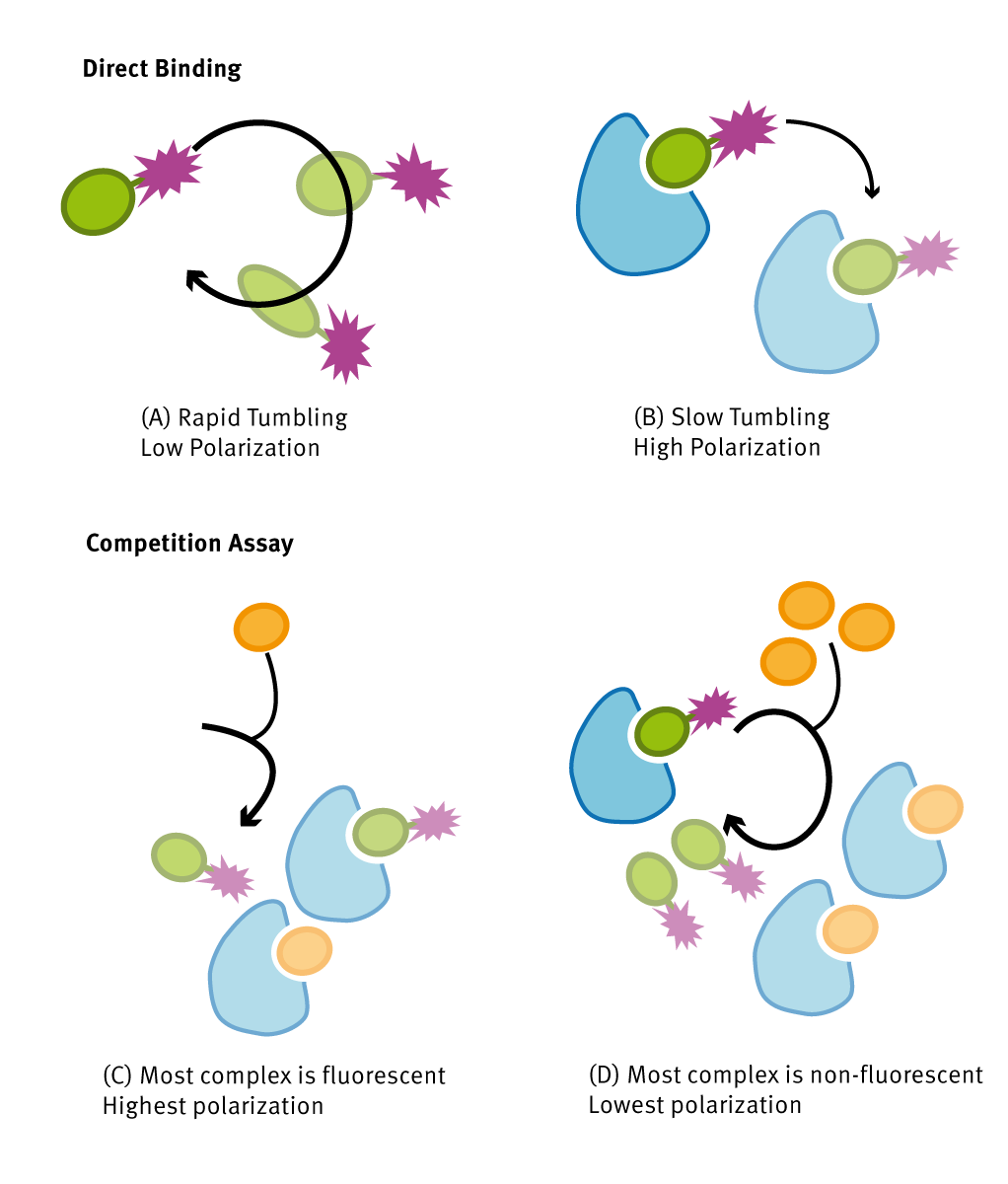
Materials & Methods
- black, 384-well, low flange microplates from Corning, UK
- TAMRA-conjugated peptides provided by NKI Protein Facility (Amsterdam)
- LC1 protein was recombinantly produced, according to reference 1
- Multi-mode microplate reader from BMG LABTECH, Germany
Experimental setup
1 nM labelled peptides were mixed to assay buffer: 15 mM KH2PO4 pH 7.2, 5% glycerol, 1 mg/ml BSA. Mix was then distributed equally across the plate. For each experiment, triplicates were prepared: starting concentration of protein LC1 was 4 μM, and then this was serially diluted 1:1 using the mix in the other wells. For competitive assays, constant enzyme concentration (around Kd) were mixed with 1 nM labelled ligand in the same assay buffer as above. Competitors were then titrated in using serial 1:1 dilutions as described for direct binding assay. The typical concentration range started from 10 μM.
(A) H3 labeled peptides in solution show a rapid rotation that causes dispersion in different direction of the incident polarized light: the resulting emitted polarization signal is low. (B) Upon binding to LC1, the high-molecular weight complex results in a slow rotation of the fluorescent molecule leading to high polarization values. (C,D) Non-tagged peptides, in orange, compete for LC1 active site. As competitor concentration increases, more freely labelled peptide remains in solution. This results in a low FP value, comparable to results in (A).
CLARIOstar instrument settings
| Detection Mode: | Fluorescence Polarization, Endpoint |
| No. of flashes: | 50 |
| Excitation: | 540-20 |
| Emission: | 590-20 |
| Target temperature: |
25°C |
Results & Discussion
We first devised a direct binding experiment using three different H3-derived peptides, all previously described as substrates and/ or inhibitors of LC1 complex. On a relative scale, results obtained are consistent with activity measured before by the working group. The difference in the peptides consisted in different lengths and/or different amino-acid compositions as listed below:
- meH3 (pep01) = H3 N-terminal tail, methylated on Lys4, 21 residues
- H3K4M (pep02) = H3 N-terminal tail, baring the mutate residue in position 4, 21 residues
- SN12 (pep03) = Transcription factor SNAIL, an H3 analogue, consisting of 12 residues (N-terminal)
As shown in figure 2, all three peptides do bind to LC1.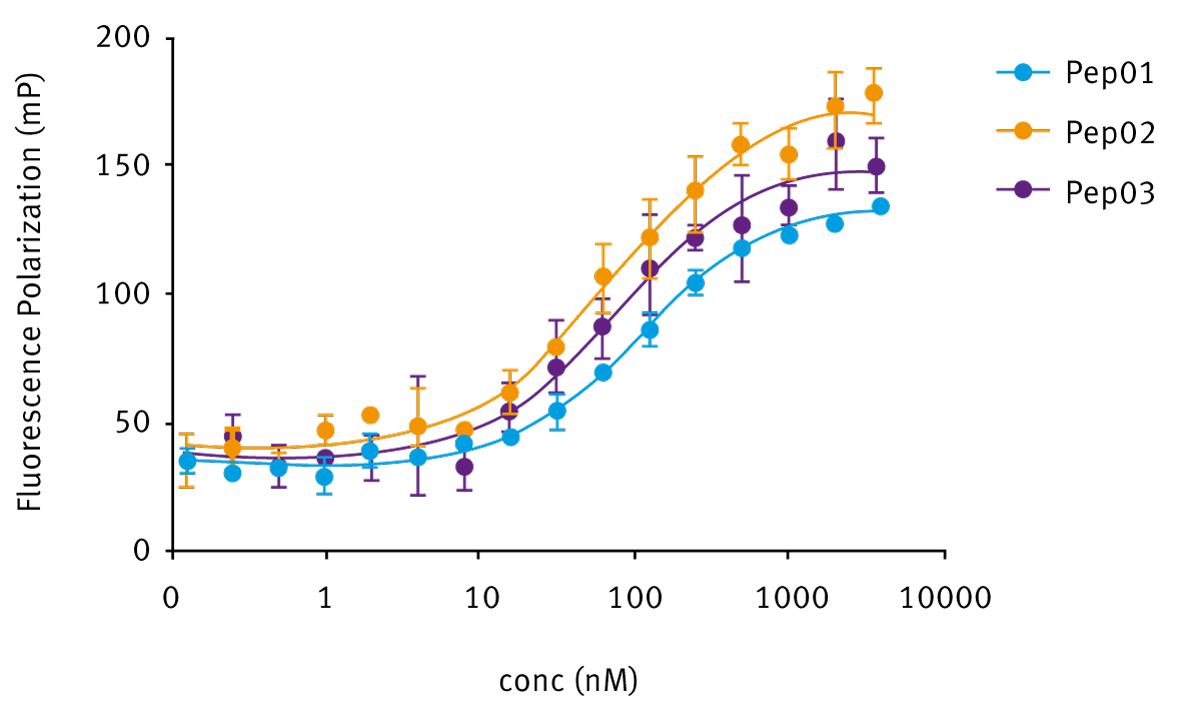 Acquired data was analysed using Mars Software and graphically edited in GraphPad. Association constants were calculated modifying the equation for fluorescent anisotropy (see equation 1) using constant label concentration as constrain.
Acquired data was analysed using Mars Software and graphically edited in GraphPad. Association constants were calculated modifying the equation for fluorescent anisotropy (see equation 1) using constant label concentration as constrain.

To further assay the binding of histone tail derivate peptides and to confirm association parameters, we proceeded using a competition approach: for this set of experiments a new test mix was prepared, as described above. We titrated in different potential competitors, using the same enzyme concentration, which depended on Kd values obtained from direct binding assays. As shown below (Fig. 3), we were able to assess affinity of different potential binders using a different approach but still in fluorescence polarization.
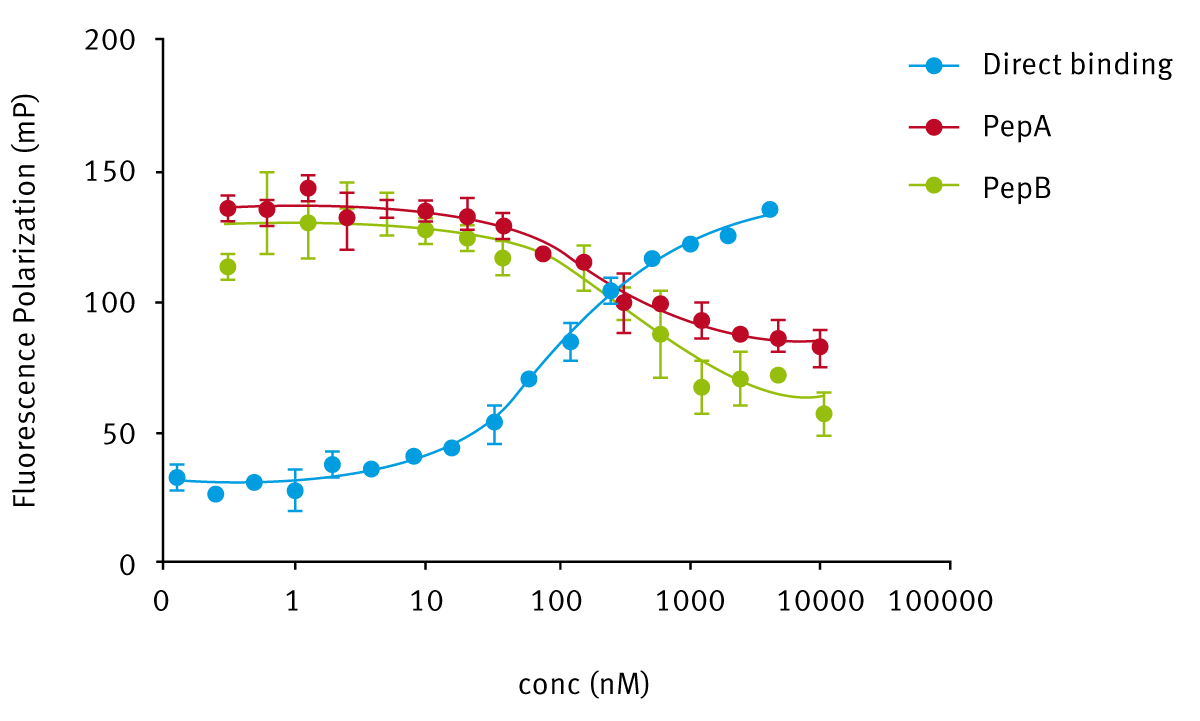
Table 1 enlists all molecules used in both direct and competitive assays with the resulting affinities values (Kd).
Table 1: Summary of association constants for LC1-peptides binding. For pep01-02-03, the different constants are to be considered coherent, as differences might be due to the sensitivity of the measurement and in the mix preparation.
| Ligand | Kd | |
| Pep01 | 107.3 nM ± 7.6 nM | Direct Binding |
| Pep02 | 76.4 nM ± 10.4 nM | Direct Binding |
| Pep03 | 81.9 nM ± 16.7 nM | Direct Binding |
| Pep A | 147.1 nM ± 35.4 nM | Competition |
| Pep B | 316.6 nM ± 59.9 nM | Competition |