
PHERAstar FSX
Lecteur de microplaques HTS puissant et ultra-sensible


En général, l'absorbance est un processus d'interaction entre la lumière et la matière. Lorsque la lumière touche un objet qui absorbe une partie de cette lumière, on parle d'absorbance. La lumière absorbée n'est pas perdue pour autant ; elle est transformée en chaleur ou en énergie chimique lorsque la molécule absorbante est excitée. L'absorbance est un processus physique que nous rencontrons tous les jours lorsque nous voyons des couleurs : la lumière blanche, comme la lumière du soleil, contient toutes les couleurs du spectre visible. Lorsqu'elle éclaire l'herbe verte, celle-ci absorbe toutes les couleurs sauf le vert. La lumière verte est alors réémise, ce qui donne à l'herbe sa couleur.

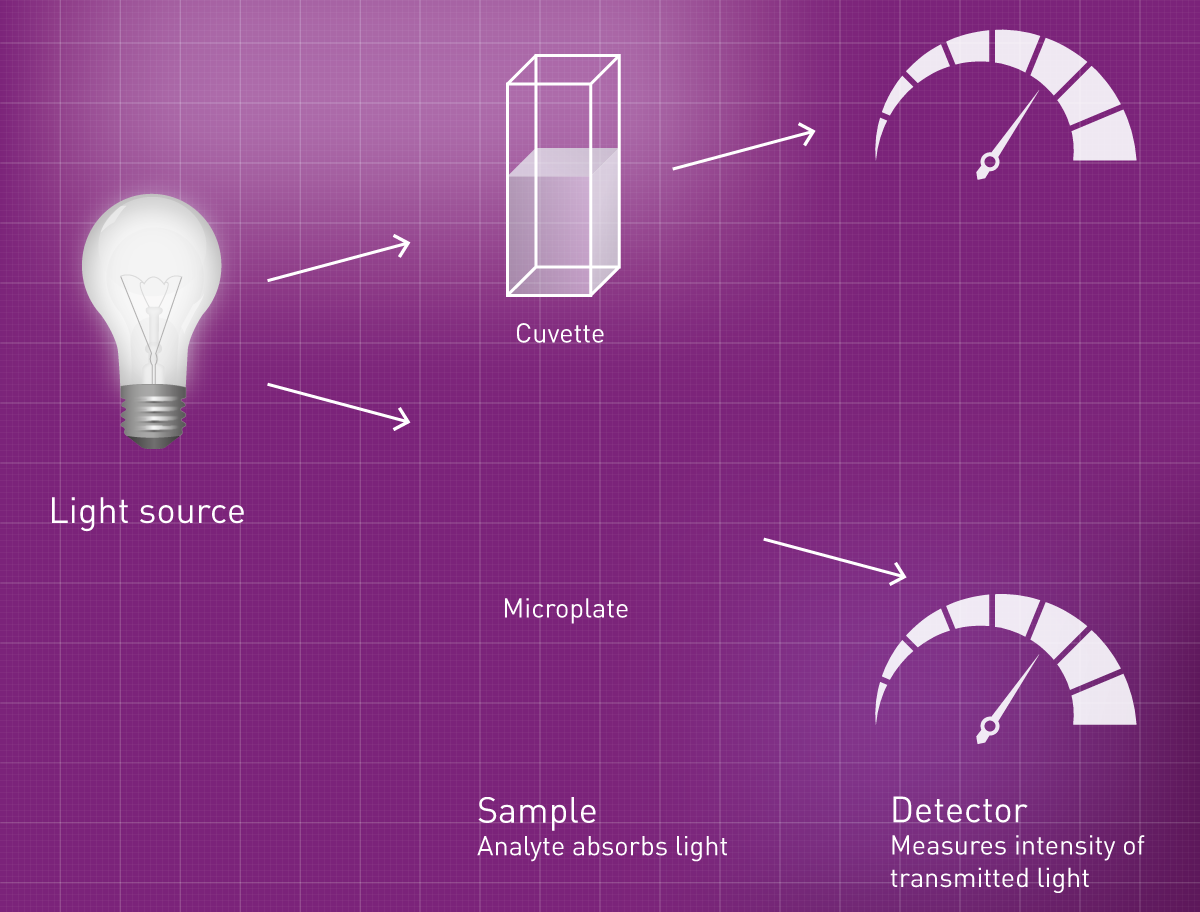
Traditionnellement, les mesures d'absorbance sont effectuées dans une cuvette : une solution contenant un analyte dont les caractéristiques d'absorption sont connues est placée dans une cuvette. Un lecteur d'absorbance détermine alors l'absorbance en envoyant une lumière d'intensité connue à travers l'échantillon, puis en détectant l'intensité de la lumière derrière l'échantillon. La lumière qui n'a pas atteint le détecteur a été soit absorbée, soit diffusée. La partie diffusée est déterminée séparément en mesurant des blancs appropriés, puis soustraite à la valeur obtenue pour obtenir l'absorbance pure de la substance d'intérêt.
La partie de la lumière qui traverse l'échantillon est appelée transmission et s'exprime principalement en pourcentage (voir la figure 2). Plus la solution contient d'analyte, plus elle absorbe de lumière et plus la transmission est faible. L'absorbance, en revanche, correspond à la partie de la lumière absorbée par l'analyte. C'est la valeur absolue du logarithme à la puissance 10 de la transmission. Voici quelques équations mathématiques et chiffres qui expliquent la transmission et l'absorbance :
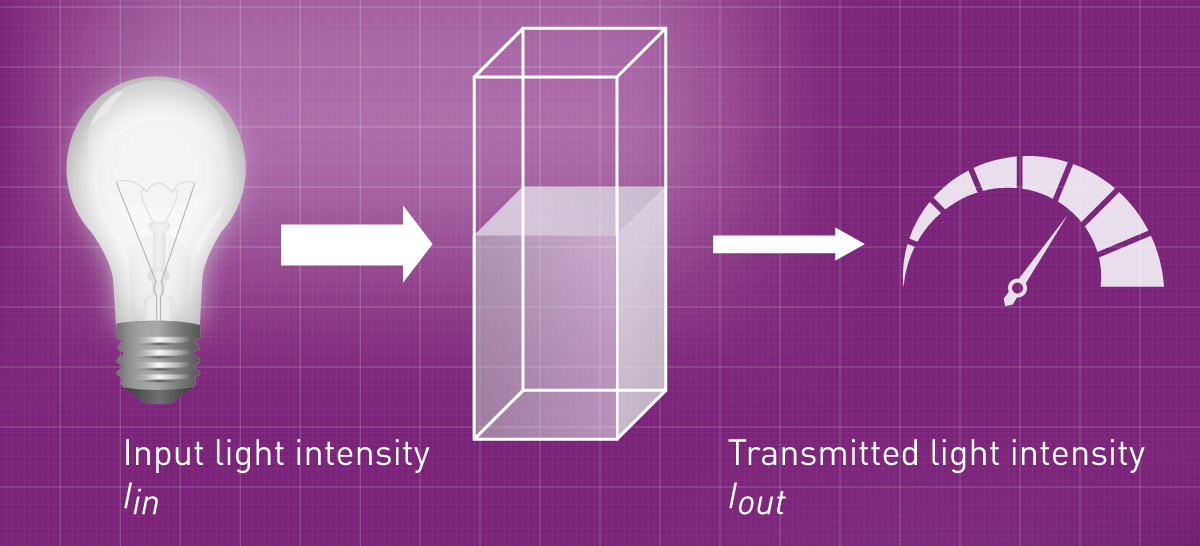
Transmission : T =Iout /Iin
Absorbance : A =-log10T
Tableau 1 : Exemples de valeurs de transmission et d'absorbance
| Transmission | Absorbance |
| 0,1 (ou 10 %) | 1 DO |
| 0,01 (ou 1 %) | 2 DO |
| 0,001 (ou 0,1 %) | 3 DO |
Après avoir effectué une mesure d'absorbance, le résultat est une valeur donnée soit en transmission, soit en densité optique. Toutefois, l'objectif étant de quantifier une substance en solution, la question se pose de savoir comment convertir le signal en valeur de concentration. Il existe deux méthodes pour y parvenir : utiliser la loi de Beer-Lambert ou mesurer une courbe standard parallèlement à des échantillons de concentrations inconnues.
La loi de Beer-Lambert décrit la relation entre l'absorbance, la longueur du trajet et la concentration d'une substance absorbante :
A = c*d*ε
Remplacé par c : c = A/(d*ε)
La loi de Beer-Lambert s'écrit : A = εcd, où A est l'absorbance, c la concentration, d la longueur du trajet et ε le coefficient d'extinction.
Elle indique que l'absorbance est proportionnelle à la concentration multipliée par la longueur du trajet et par le coefficient d'extinction2. La longueur du trajet correspond à la distance que la lumière doit parcourir pour traverser l'échantillon. Dans une cuvette, par exemple, ce trajet est normalisé à 1 cm. Le coefficient d'extinction est une constante spécifique à une substance absorbante et à une longueur d'onde donnée, correspondant généralement au maximum d'absorbance de la substance. Il fournit des informations sur la force avec laquelle la substance absorbe la lumière à une longueur d'onde spécifique. À titre d'exemple, le coefficient d'extinction de masse de l'albumine sérique bovine (BSA) est de 0,67 µl*cm-1*µg-1. Par conséquent, une solution de 1 µg/µl de BSA avec une longueur de trajet de 1 cm a une absorbance de 0,67 DO.
La loi de Beer-Lambert est très utile, car elle permet de quantifier les substances absorbantes sans qu'il soit nécessaire d'ajouter d'autres réactifs. Cependant, elle présente certaines limites, comme indiqué ci-dessous.
La loi de Beer-Lambert ne peut être utilisée que si :
Si l'un de ces critères ne s'applique pas, il est possible de mesurer les analytes de manière indirecte et/ou d'utiliser une courbe standard. Les tests colorimétriques de quantification des protéines, tels que le test de Bradford, dépendent par exemple d'une substance qui augmente l'absorbance en présence de protéines. L'augmentation est mesurée dans une courbe standard contenant des concentrations connues de protéines, puis dans des échantillons, ce qui permet de calculer leur concentration.
La mesure de la transmission/absorption de la lumière nécessite d'abord de la lumière. Différentes sources lumineuses peuvent être utilisées pour effectuer des mesures d'absorbance. Celles-ci diffèrent par la gamme spectrale qu'elles couvrent, par leur intensité et par la stabilité de la lumière qu'elles émettent.
Les lampes halogènes au tungstène couvrent la plage de 360 nm à plus de 1000 nm et sont souvent utilisées en raison de leur rentabilité. Les lampes flash au xénon, quant à elles, couvrent la région spectrale de 220 à 1000 nm et la gamme UV du spectre. Elles permettent ainsi de détecter et de quantifier les acides nucléiques (260 nm) et les protéines (280 nm). Les lecteurs de microplaques par absorbance de la marque BMG LABTECH sont équipés d'une lampe flash au xénon pour les mesures d'absorbance, ce qui leur confère une flexibilité maximale pour mesurer n'importe quelle longueur d'onde comprise entre 220 et 1000 nm, ou l'ensemble du spectre.
Les liquides d'intérêt sont transférés dans une cuvette ou une microplaque afin de mesurer leur absorbance. Le matériau de la cuvette et de la microplaque est toujours transparent afin d'assurer une transmission maximale de la lumière, car c'est l'absorbance de la solution qui intéresse le chercheur, et non celle du matériau. Cependant, divers matériaux sont utilisés et diffèrent d'un matériau à l'autre. Le polystyrène est le matériau le plus souvent utilisé. Il peut être modifié pour présenter des caractéristiques de liaison spécifiques, que ce soit pour la culture cellulaire ou les applications ELISA. Cependant, ils ne sont pas transmissifs dans la gamme des ultraviolets, ce qui nécessite l'utilisation de microplaques spéciales en copolymère d'oléfine cyclique pour mesurer l'absorbance en dessous de 400 nm.
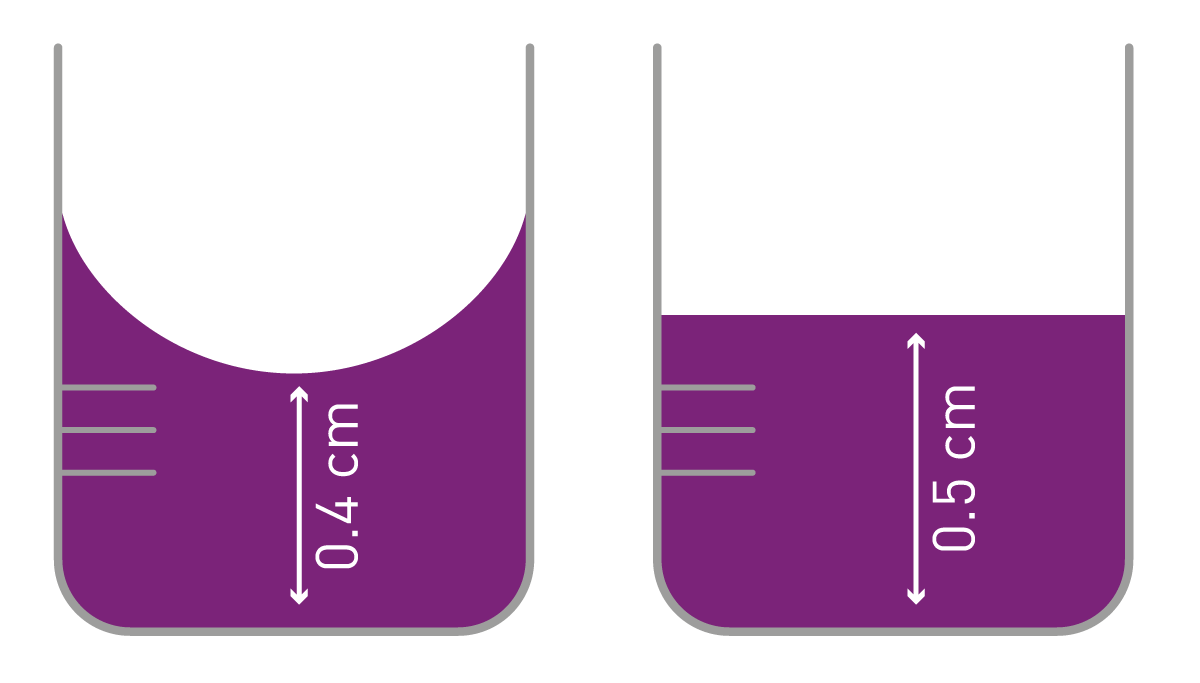
Un deuxième aspect à prendre en compte est le volume d'échantillon nécessaire pour obtenir des mesures d'absorbance fiables. Les cuvettes standard nécessitent environ 4,5 ml, tandis que les cuvettes semi-micro permettent de réduire ce volume à 1,5 ml et que les cuvettes micro-volume se contentent de 70 µl. La mesure en cuvette est horizontale : la lumière est dirigée d'un côté vers l'échantillon et la lumière transmise est détectée de l'autre côté. La géométrie des cuvettes permet donc d'obtenir une longueur de trajet normalisée de 1 cm. C'est l'une des principales différences avec les mesures en microplaques, qui se déroulent verticalement (de haut en bas ou de bas en haut). La longueur du trajet varie donc également en fonction du volume de l'échantillon et il est nécessaire d'utiliser des volumes égaux pour réaliser une expérience. En outre, les effets de ménisque jouent un rôle important dans la longueur de trajet lors des mesures d'absorbance en microplaques, comme expliqué en détail dans la publication : « Comment traiter la longueur de trajet et le ménisque dans les microplaques ». Les mesures effectuées au milieu d'un puits de microplaque ont un trajet beaucoup plus court en présence d'un ménisque important qu'en l'absence de ménisque. Ceci est particulièrement important lors de l'application de la méthode de Beer-Lambert ou lorsque les ménisques varient fortement au cours d'une expérience. Dans les solutions aqueuses, il est possible de corriger les changements de longueur de trajet dus au ménisque en exploitant l'absorbance de l'eau (correction de la longueur de trajet du pic de l'eau).
Des microplaques standard à 96 puits sont généralement utilisées avec des volumes d'échantillons compris entre 100 et 300 µl, ce qui correspond à une longueur de trajet d'environ 2,9 à 7,4 mm. Il peut toutefois être nécessaire de réduire encore le volume de l'échantillon sans compromettre la longueur du trajet. À cette fin, il est possible d'utiliser des microplaques à demi-surface ou à densité plus élevée. Ces microplaques offrent une surface de puits plus petite, mais la même hauteur que les microplaques standard à 96 puits, ce qui permet d'obtenir des longueurs de trajet élevées avec des volumes plus faibles.
Afin de corriger les mesures d'absorbance indésirables provenant des composants du tampon, de la microplaque et des effets de diffusion de la lumière, on mesure un blanc parallèlement aux échantillons. Le blanc approprié contient tous les composants de l'essai, à l'exception de l'analyte. Inversement, un blanc constitué de PBS ou d'eau est souvent insuffisant.
Il faut accorder une attention particulière aux particules présentes dans les échantillons analysés et dans les blancs. En effet, les particules diffusent la lumière et augmentent la valeur d'absorbance mesurée, car moins de lumière atteint le détecteur. Lorsqu'elles se déplacent dans la solution, elles sont détectées lorsqu'elles bloquent le trajet de la lumière. Lorsqu'elles se déplacent hors du chemin de détection, elles ne sont pas mesurées. Ce phénomène augmente la variabilité des données d'absorbance. Si les particules ne peuvent pas être éliminées des solutions à mesurer, il est recommandé de couvrir une plus grande surface du puits de la microplaque lors de la mesure d'absorbance, une caractéristique que tous les appareils BMG LABTECH proposent pour ce type de mesure.
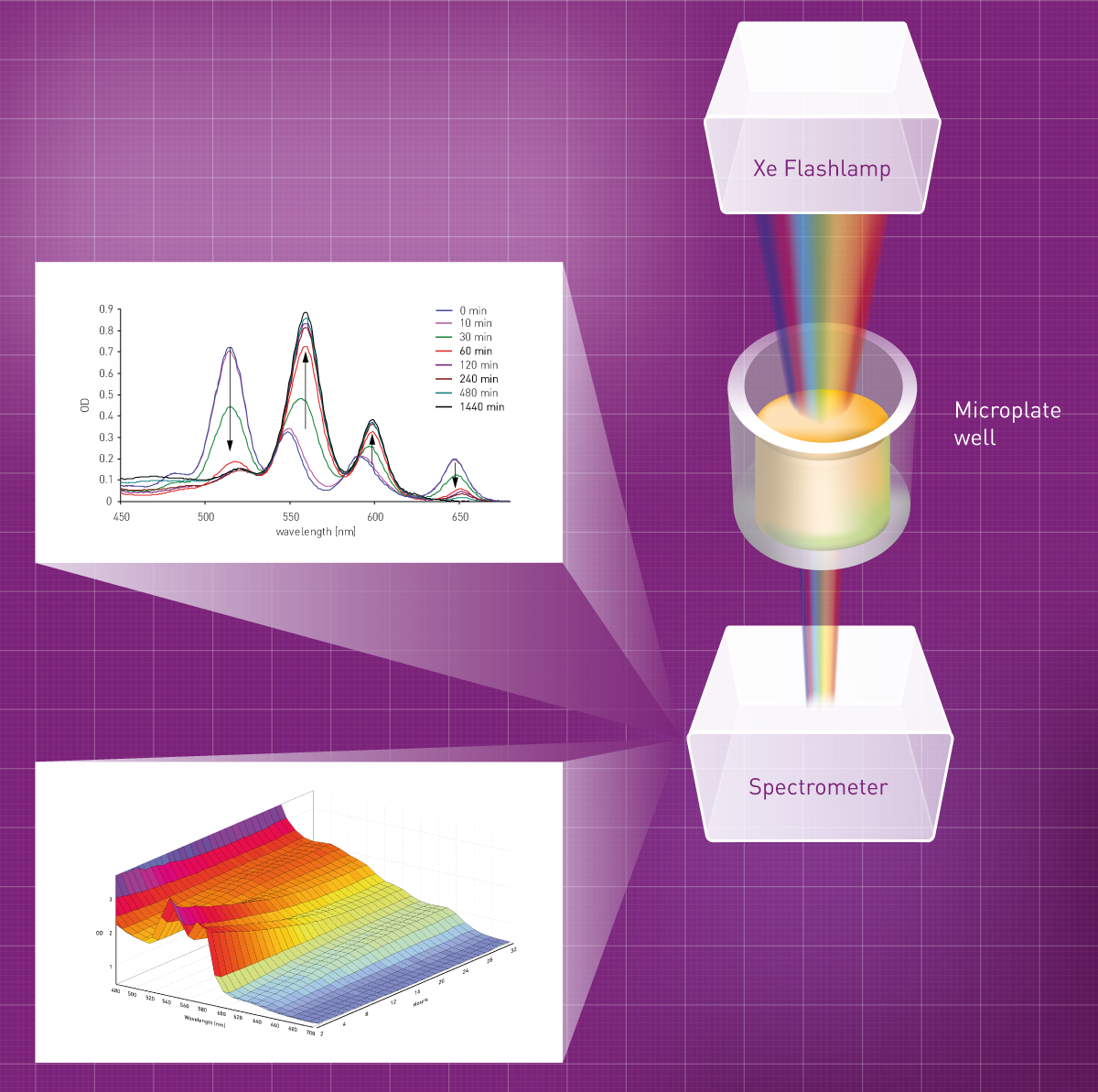
Une fois la lumière transmise par l'échantillon, il faut la détecter. Deux types de détecteurs sont couramment utilisés dans les lecteurs de microplaques : un tube photomultiplicateur (PMT) ou un spectromètre à charge couplée (CCD). Les systèmes basés sur un PMT nécessitent de sélectionner la longueur d'onde avant la détection. Pour ce faire, des filtres optiques ou des monochromateurs sélectionnent la longueur d'onde souhaitée, puis la guident vers l'échantillon. La lumière transmise de la longueur d'onde souhaitée atteint alors le PMT. Le PMT amplifie le signal de la lumière transmise et produit une tension indiquant son intensité. Pour balayer l'absorbance sur une gamme de longueurs d'onde, les systèmes basés sur un PMT utilisent un monochromateur qui sélectionne la longueur d'onde souhaitée pour chaque point du spectre et la mesure de manière séquentielle.
C'est la principale différence avec le deuxième type de détecteurs d'absorbance : le spectromètre. Ce dernier divise la lumière transmise en différentes longueurs d'onde, qui sont dirigées vers un détecteur CCD permettant de capturer l'intensité de toutes les longueurs d'onde simultanément. De cette manière, les spectres ainsi que les mesures d'une seule ou de quelques longueurs d'onde peuvent être obtenus en un temps similaire. Par exemple, les instruments BMG LABTECH utilisent des spectromètres UV/vis pour les mesures d'absorbance, qui capturent un spectre entier (220-1000 nm) en moins d'une seconde.
Lecteur de microplaques HTS puissant et ultra-sensible
Lecteur de microplaques le plus flexible pour le développement de tests
Lecteur de microplaques flexible avec flux de travail simplifiés
Série de lecteurs de microplaques monomodes et multimodes évolutifs
Lecteur de microplaques par absorbance avec port pour cuvette



