Omega Series
Upgradeable single and multi-mode microplate reader series
Prion research focusses on the study of neurodegenerative diseases caused by the prion protein. Prion diseases are caused by abnormally folded, aggregating and infectious prion proteins. Also known as Transmissible Spongiform Encephalopathies (TSEs), prion diseases are rare, progressive and fatal neurodegenerative diseases that affect the brain of animals and humans.
Prion diseases include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) or "mad cow disease” in cattle, chronic wasting disease (CWD) in deer, elk, and other cervids, and scrapie in sheep.
Prion research focusses on understanding the function of the prion protein and the mechanisms underlying disease development to both establish diagnostic tests and effective therapies against these neurodegenerative diseases.
Prion diseases are a related group of neurodegenerative diseases affecting the brain both in humans and in animals. Prion diseases can spread from animal to animal, animal to human and from human to human (CJD variant). Prion research is of high relevance since much about prion diseases is still unknown, diagnoses remain complicated and there are no effective treatment options available yet, making prion diseases always fatal.
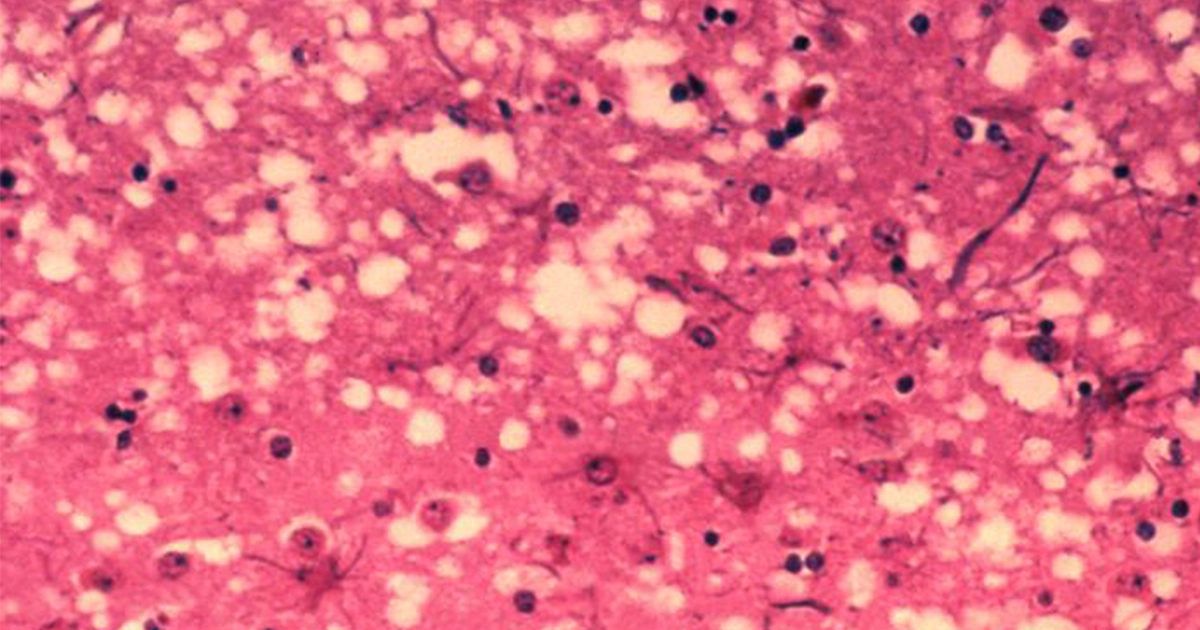
In prion diseases, the wild-type prion protein becomes abnormally folded and protein clumps accumulate in the brain. Prion protein aggregates cause neuronal death and spongiform changes in the brain (holes in the tissue with vacuole formation in neurons). In humans, the symptoms of this neurodegenerative disease are progressive and typically include memory impairment, personality changes, and motor dysfunction.
 Prion diseases are related to other neurodegenerative diseases caused by protein aggregation in the brain such as Parkinson’s disease, Alzheimer’s disease, dementia, and diseases associated to misfolding of the tau protein.
Prion diseases are related to other neurodegenerative diseases caused by protein aggregation in the brain such as Parkinson’s disease, Alzheimer’s disease, dementia, and diseases associated to misfolding of the tau protein.
The term prion was coined by Stanley Prusiner in 1982 and derives from “proteinaceous infectious particle”.1 The term refers to the at the time “heretical” hypothesis that the infectious agent could be a protein, despite the fact that all at the time known pathogens relied on some form of nucleic acid to reproduce and propagate.
Wild-type prion protein
The non-infectious form of the prion protein is called PrPC (Prion Protein Cellular isoform). PrPC is a highly conserved cell surface protein with a molecular mass of around 35 kilodalton, containing three α-helixes at the C-terminal. It is soluble in detergents and can be digested by proteinases.2
The physiological prion protein is found in healthy animals and humans, and is neither pathogenic nor infectious. Its function is not fully clarified yet and still investigated. Current research suggests that the prion protein plays an important role in cell-cell adhesion and intracellular signalling in vivo.3
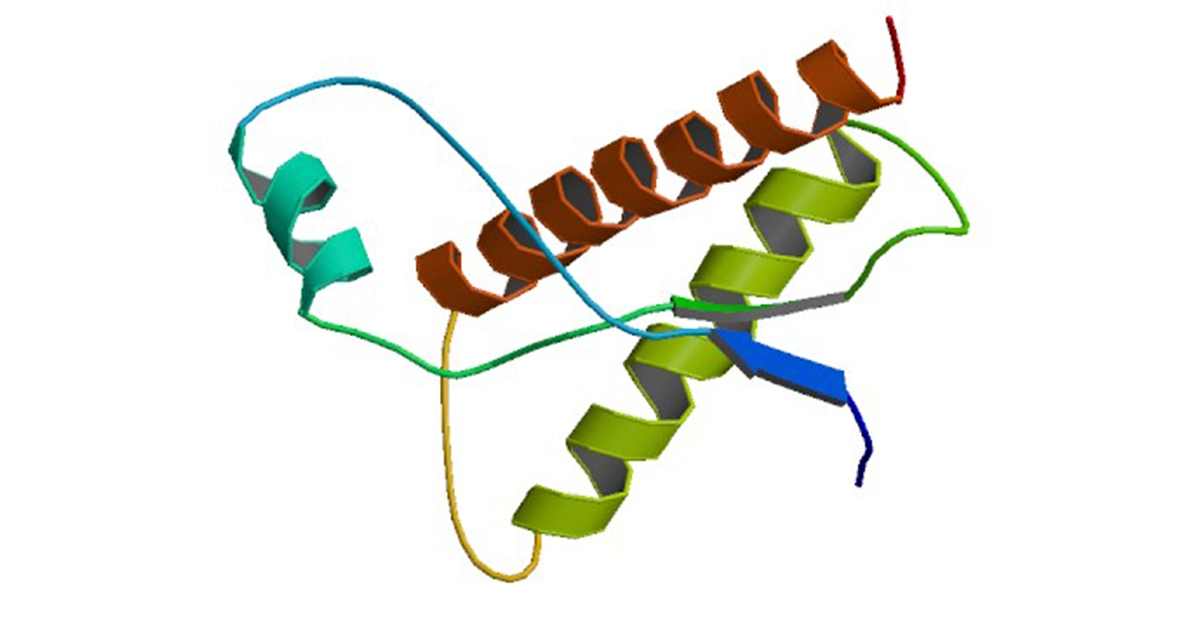 Pathogenic prion protein
Pathogenic prion protein
In the pathogenic and infectious prion isoform PrPSc (Prion Protein Scrapie, the sheep prion disease), the primary sequence is identical to the wild-type PrPC prion isoform but with a different secondary and tertiary structure. The PrPSc prion isoform undergoes a conformational rearrangement that results in a β-sheet-rich structure and has a high propensity to aggregate and build detergent-insoluble, highly structured polymeric amyloid fibres which accumulate in the brain. As the PrPSc prion isoform is resistant to protein digestion by proteases, it is also often referred to as PrPRES.
The causative molecular events that induce the formation of the PrPSc prion protein isoform and trigger neurodegenerative events in the brain remain poorly understood.
The current scientific view is that prion proteins spread by self-propagation of the infectious misfolded PrPSc prion protein isoform. The most widely accepted model that explains the prion replication is the “protein only” hypothesis, where the conversion of wild-type PrPC prion protein to the pathological PrPSc prion protein isoform is a post-translational conformational change from a largely α-helix-rich structure to a ß-sheet-rich conformation.
It is generally accepted that the infectious prion protein isoform PrPSc serves as a template that, when bound to the physiologic PrPC prion isoform, promotes its conformational change to PrPSc in a seeded polymerization reaction. The newly formed PrPSc prion proteins act as additional templates and propagate the conversion of more wild-type prion proteins, leading to an exponential accumulation of PrPSc prion isoforms in the brain.
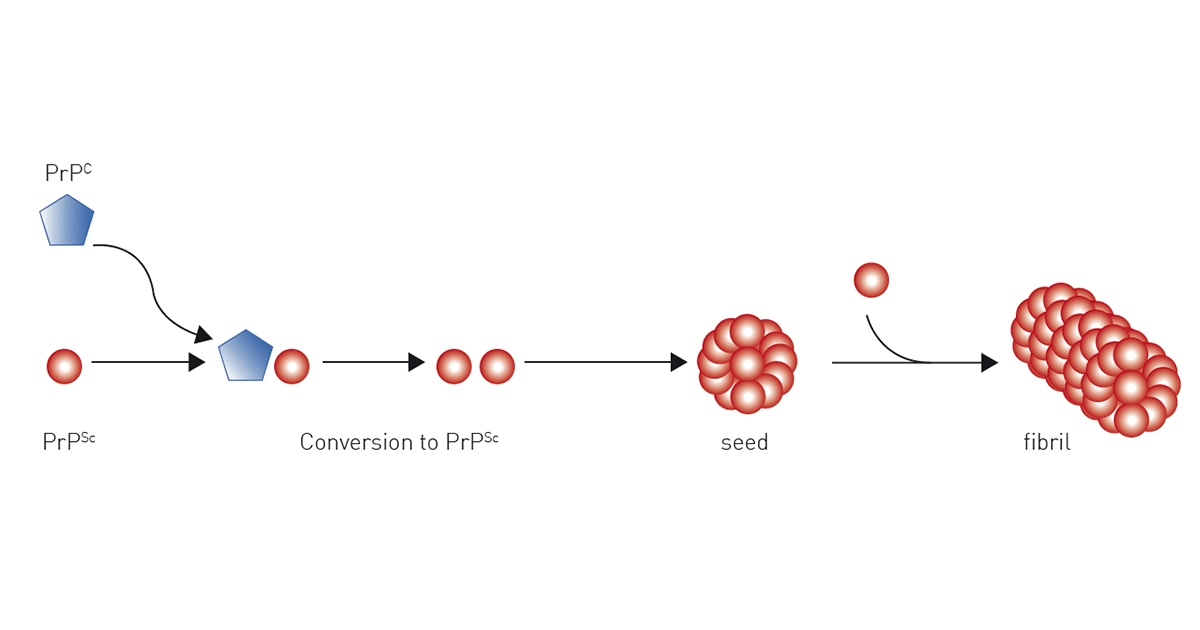
PrPSc prion proteins polymerise in tightly packed β-sheets to form amyloid fibrils on which unbound PrPSc prion proteins attach, allowing the fibre to grow. Fibres “replicate” when rupture induces growing end multiplication. 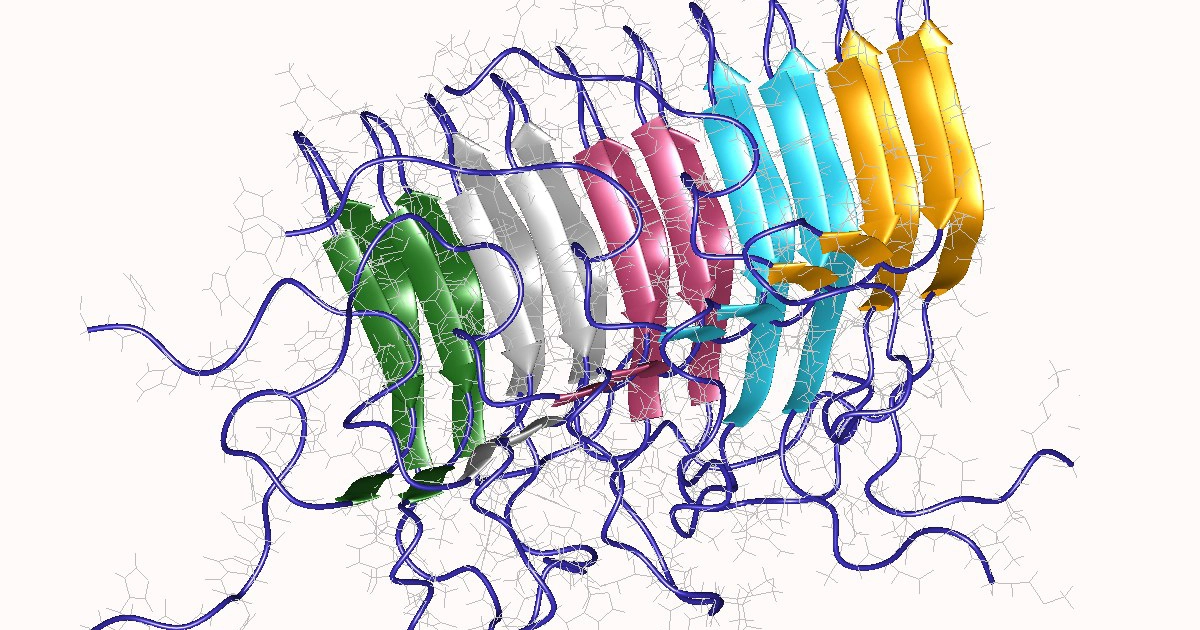
Amyloids are protein aggregates characterised by a fibrillar morphology and β-sheet secondary structure that have been linked to various diseases. They typically form fibrous deposits (plaques) around cells which disrupt tissue and organ function.
These fibres are thought to interfere with synapse function in the brain and cause neuronal cell death, eventually resulting in the neurodegenerative symptomatology associated to Creutzfeldt-Jakob disease, "mad cow disease”, chronic wasting disease (CWD), and scrapie.
However, the underlying mechanism of disease still remains unclear to scientists. The main focus of prion research is nowadays trying to understand how cells in the brain interact with prion proteins and how these interactions affect neurodegenerative disease progression.
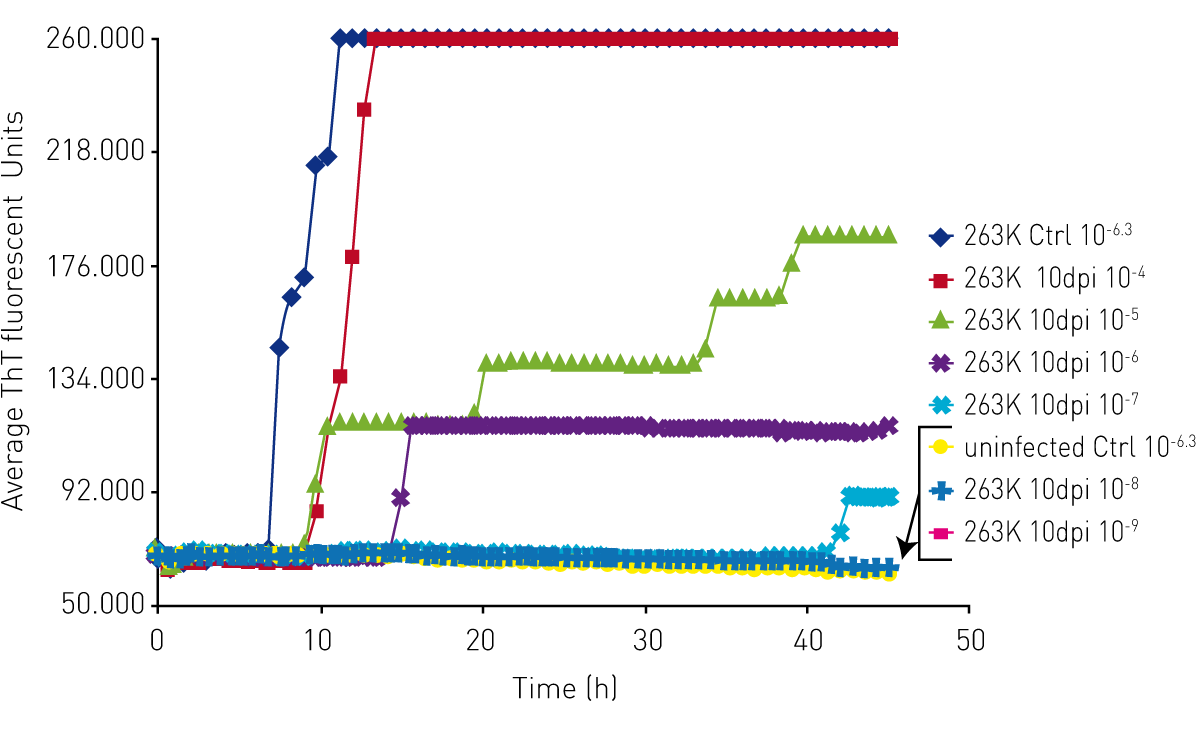
Initially, prion diseases could only be diagnosed by taking a biopsy of the brain tissue, typically post mortem. Scientists at the Rocky Mountain Laboratories in Hamilton, Montana, USA have developed a rapid and sensitive diagnostic test for prions called the Real Time-Quaking Induced Conversion (RT-QuIC) assay. Today, RT-QuIC is used at different facilities throughout the world to diagnose human prion disease such as CJD but also for animal prion diseases like scrapie, mad cow and CWD.
RT-QuIC uses a cell-free conversion reaction in which recombinant PrPC prion proteins are converted by an original seed into the PrPSc prion isoform, aggregate to exponentially amplify and enable detection of otherwise undetectable prion levels. RT-QuIC assays use fluorescence intensity detection of Thioflavin T and cycled shaking to produce prion protein multimers when a PrPSc prion seed is present.4
Today, the prion RT‐QuIC assay is the most widely used assay for prion disease diagnostics in patients as it allows an accurate ante mortem diagnosis of Creutzfeldt-Jakob disease using cerebrospinal fluid 5 or brushings of the olfactory mucosa. The latter nasal brush test for human prion detection can rapidly and accurately diagnose Creutzfeldt-Jakob disease (CJD) in living patients. 6
Besides prion diagnostics, RT-QuIC is also used to test compounds and identify candidate molecules that inhibit prion protein formation or aggregation. Researchers are also working to adapt the RT-QuIC prion assay to other neurodegenerative diseases, to detect the proteins that cause Alzheimer’s disease, Parkinson’s disease, dementia, and other neurological diseases that involve protein misfolding.
Although the search for pharmacological treatments for prion diseases started more than 30 years ago, no validated therapy for livestock or human prion diseases is available yet.
An initial strategy has been to screen for molecules that can inhibit the formation of the pathological prion protein isoforms. In fact, it has been shown that such compounds can extend the lives of experimental animals inoculated with prions. However, the therapeutic efficacy of such inhibitors against neurodegenerative brain damage has so far been limited by toxicity and insufficient bioavailability to the central nervous system.
The RT-QuIC prion assay relies on a microplate reader for signal quantification, as well as for shaking and temperature conditions conducive for seeding and formation of aggregates.
BMG LABTECH microplate readers play a central role in prion research, since the RT-QuIC assay has been originally developed on the FLUOstar® Omega. This reader has become the worldwide standard for prion detection for a variety of species, including human prions from various tissues.
The RT-QuIC prion assay takes advantage of the solid construction of BMG LABTECH microplate readers to perform cycles of shaking on and off for an extended time, while periodically reading the plate to monitor changes in fluorescent signal as Thioflavin T is incorporated into prion aggregates.
For more details on running the RT-QuIC assay on the FLUOstar Omega, have a look at the application note Real-time quaking induced conversion assay for prion seeding.
Moreover, in the scientific talk “Optimizing sensitivity and specificity in the RT- QuIC assay” Dr. Davin Henderson from Colorado State University discusses amyloid amplification and analyses important factors to be considered for prion detection with the RT-QuIC assay.
Other microplate-based application for prion research
Besides RT-QuIC, microplate readers can be used for different prion applications. For instance, the Conformational Dependent Immunoassay (CDI) takes advantage of the epitope specificity of monoclonal antibodies and the fact that different prion forms contain distinct epitopes that can be recognized by these antibodies. Using two antibodies designed to specifically recognize two different prion epitopes, the CDI becomes a fairly straightforward sandwich immunoassay in which samples are bound to plates using the capture antibody and subsequently incubated with detection antibody. Detection, in this case, is achieved by labelling the antibody with a lanthanide to enable time-resolved fluorescence detection on a plate reader.7
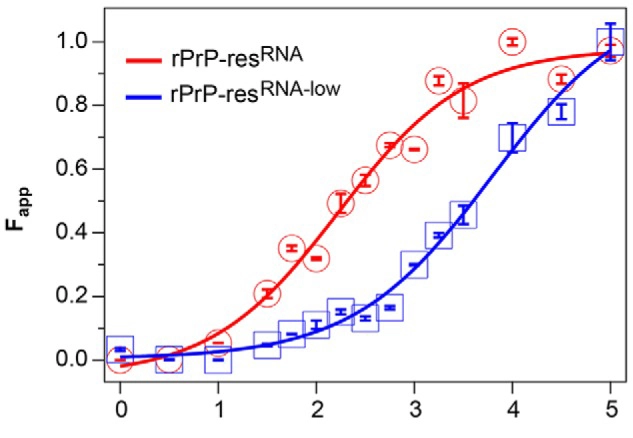
Another example is the bioluminescent prion assay (BPA), an assay based on Gaussia luciferase (GLuc) that was used to look at the progressive binding of prion proteins, focusing on the initial dimerisation event. In BPA, 2 prion forms are expressed in RK13 cells. One form was tagged with the N-terminal portion of GLuc, the other with its C-terminal portion. When the 2 prion forms are brought together by the dimerisation event the 2 fragments of GLuc are also brought into proximity. The result is a reconstituted, fully active GLuc protein that, in the presence of the appropriate substrate coelenterazine, can now produce a signal that can be detected by luminescence on a plate reader. Researchers used this assay to develop a compound screen to identify small molecules that inhibit prion dimerisation. 8
Prion diseases share common pathogenic features with other neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and diseases related to the tau protein. In fact, these non-infectious neurodegenerative diseases are associated with the presence of misfolded protein deposits, the accumulation of amyloid fibres and progressive neuronal damage in the brain.
α-synuclein, amyloid-β and tau, the misfolded proteins involved in these diseases, all share key common structural, biophysical, and biochemical characteristics with the prion protein. Moreover, aggregation and self-propagation of misfolded proteins in these diseases seem to happen through mechanisms comparable to prions. Accordingly, it has been suggested that all these disorders should be considered as belonging to the prion like group of diseases.
Microplate readers can also be used to gain further insight in these neurodegenerative diseases.
Thioflavin T-based detection of protein aggregation has been successfully employed to analyse aggregation of amyloid-β for Alzheimer’s disease: “Following Abeta fibrillization/aggregation in real-time using a FLUOstar Omega microplate reader”.
As an alternative to Thioflavin T-based aggregation assays, prefibrillar species of amyloidogenic proteins can be monitored by fluorescence polarization using the fluorescent probe TPE-TPP, as described in the application note “Novel aggregation-specific fluorogen monitors prefibrillar protein aggregation by fluorescence polarisation”.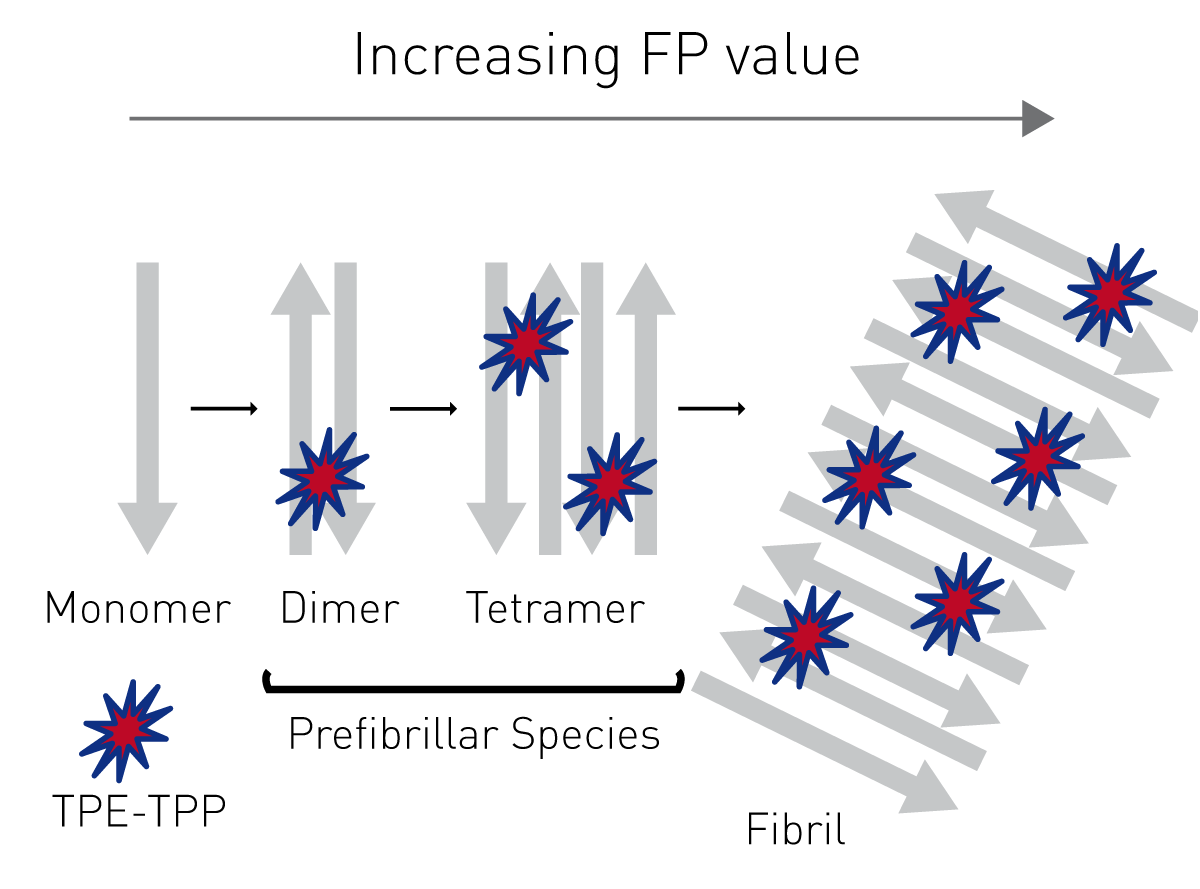
Tau aggregation can be analysed by a time-resolved FRET immunoassay, as shown in the application note “Detection of human tau protein aggregation”. Moreover, tau, together with other Alzheimer’s disease targets can be easily quantified by ELISA as shown in the application note “Fast and accurate detection of Alzheimer’s Disease targets with SimpleStep ELISA kits and SPECTROstar Nano”. and “Comparing site specific phospho-tau in normal and Alzheimer’s disease brains”.
Are you interested in more neurobiology content? Check out our neurobiology research area section. Here you can find other blog posts, webinars, references on peer-reviewed papers and application notes covering this topic.
Upgradeable single and multi-mode microplate reader series
Neurodegenerative disease ultimately leads to the death of neurons as neuronal functions deteriorate. Find out how microplate readers can be used to study neuronal cell death and its link to neurodegenerative disease.
The disruption of mitochondrial function is linked to neurodegenerative diseases. Find out how microplate readers advance research into mitochondrial dysfunction and neurodegenerative diseases.
The way proteins misfold and aggregate is linked to many neurodegenerative diseases. Find out how microplate readers can help advance research into protein misfolding.
Alpha-synuclein is a key protein involved in neurodegenerative diseases like Parkinson’s. Find out how microplate readers can help advance alpha-synuclein research.
Amyloids are thought to play a crucial role in neurodegeneration. Find out how microplate readers help advance amyloid research.
The tau protein plays a role in many neurological diseases and disorders. Find out about neuronal toxicity induced by tau and how microplate readers can aid tau research.