Introduction
GTP-binding proteins (G-proteins) are important, well-described cellular signaling molecules. Heterotrimeric G-proteins are composed of three subunits Gα, Gβ, and Gγ, and are typically bound to seven trans-membrane G-protein coupled receptors (GPCRs). The Gα-subunit binds guanine nucleotides while the Gβ and Gγ subunits form an obligate heterodimer. In its inactive state, the GDP bound Gα subunit is bound to Gβγ. Upon agonist activation the receptor acts as a guanine nucleotide exchange factor (GEF), resulting in the release of GDP and subsequent binding of GTP. The binding of GTP causes a dramatic conformational change in three flexible switch regions of Gα (Fig. 1 dark red and dark blue) resulting in the dissociation of Gα-GTP from Gβγ.
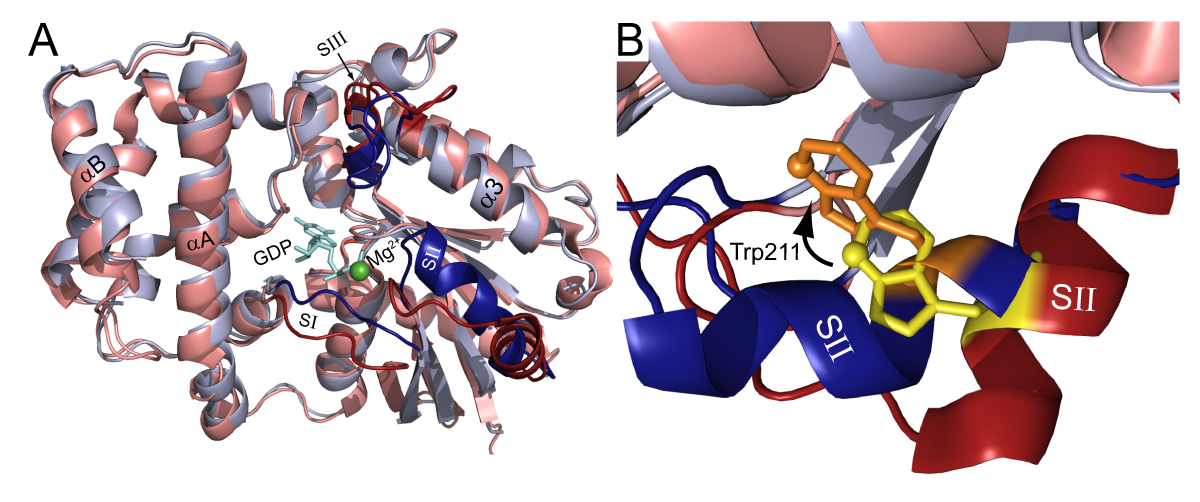
The duration of activation is controlled by the hydrolysis rate of GTP. Two well-described accessory protein families affect the kinetics of Gα subunits by either accelerating GTP hydrolysis (the RGS proteins) or retarding GDP release (the GoLoco proteins). Regulators of G-protein signaling rapidly accelerate the GTP hydrolysis of Gα subunits by stabilizing the transition state; while GoLoco motifs act as GDIs (guanine nucleotide dissociation inhibitors), preventing GDP dissociation by adding a second arginine side-chain to the contacts made to the bound nucleotide.
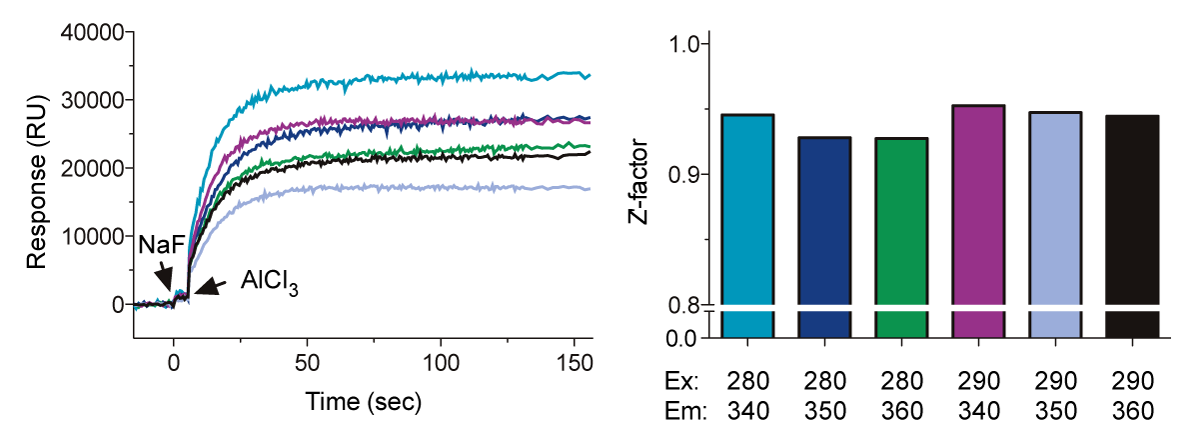
In this application note, we describe the use of the BMG LABTECH’s microplate reader to monitor changes in the intrinsic fluorescence of a highly-conserved tryptophan located in the switch II region of Gα subunits (Fig. 1, “SII”). The conformational change in SII decreases the exposure of the Trp residue to the aqueous environment, resulting in an increase in the quantum yield. One can quantify this event by measuring the increase in Gα protein fluorescence at 350 nm upon excitation at 280 nm. In this application note, we have optimized the assay by varying concentrations of Gα, changing assay buffers, and shifting excitation and emission wavelengths.
Materials & Methods
All experiments were conducted at ambient temperature using Corning Black Polystyrene 96-well plates. Gαi1 was purified exactly as previously described and diluted to 1 μM in assay buffer (unless otherwise noted) and plated at an initial volume of 187 μL/well. Experiments were conducted using a 280 ± 5 nm and 350 ± 5 nm filter for excitation and emission, respectively unless specified otherwise.
To maximize data acquisition during the experiment, typical data collection was divided into three distinct phases – baseline (-15 - 0 s), activation (0 - 132 s), and plateau phase (132 - 158 s). Data were collected at 1, 0.6, and 2 s intervals for baseline, activation, and plateau phases, respectively, using the fast kinetics (well mode) function on the Omega. At 0 s, 8 μL of 0.5 M NaF and 5 μL of 1.2 mM AlCl3 were injected sequentially with a 5 s delay. NaF and AlCl3 undergo a chemical reaction to form AlF4¯, which mimics the leaving phosphate group upon hydrolysis of GTP. This stable complex, Gαi1 · GDP · AlF4¯, mimics the active, GTP-bound state of Gαi1. The gain was set to 50% relative to 200 μL of pre-activated Gαi1 · GDP · AlF4¯ to avoid saturating the signal. The previously described GoLoco motif GDI peptide, AGS3Con, was used and shown to inhibit the formation of Gαi1 · GDP · AlF4¯.
Buffers
Phosphate assay buffer (pH 8.0) - 100 mM NaCl, 100 μM EDTA, 2 mM MgCl2, 2 μM GDP, 20 mM K2HPO4/ KH2PO4 pH 8.0
HEPES assay buffer (pH 8.0) - 100 mM NaCl, 100 μM EDTA, 2 mM MgCl2, 2 μM GDP, 20 mM HEPES
Tris assay buffer (pH 8.0) - 100 mM NaCl, 100 μM EDTA, 2 mM MgCl2, 2 μM GDP, 20 mM Tris
Instrument Settings
Fluorescence Intensity - Well Mode
Keep default settings except for the following:
No. of kinetic windows - 3
Baseline
No. of intervals - 15, No. of flashes - 10, Interval time - 1 sec
Activation
No. of intervals - 220, No. of flashes - 10, Interval time - 0.6 sec
Plateau
No. of intervals - 13, No. of flashes - 10, Interval time - 2 sec
Injection - use 320 μL/s and keep smart injection unchecked
Pump 1 inject 8 μL at start time 15 s (at t=0 in the graphs)
Pump 2 inject 5 μL at start time 20 s (at t =5s in the graphs)
Results & Discussion
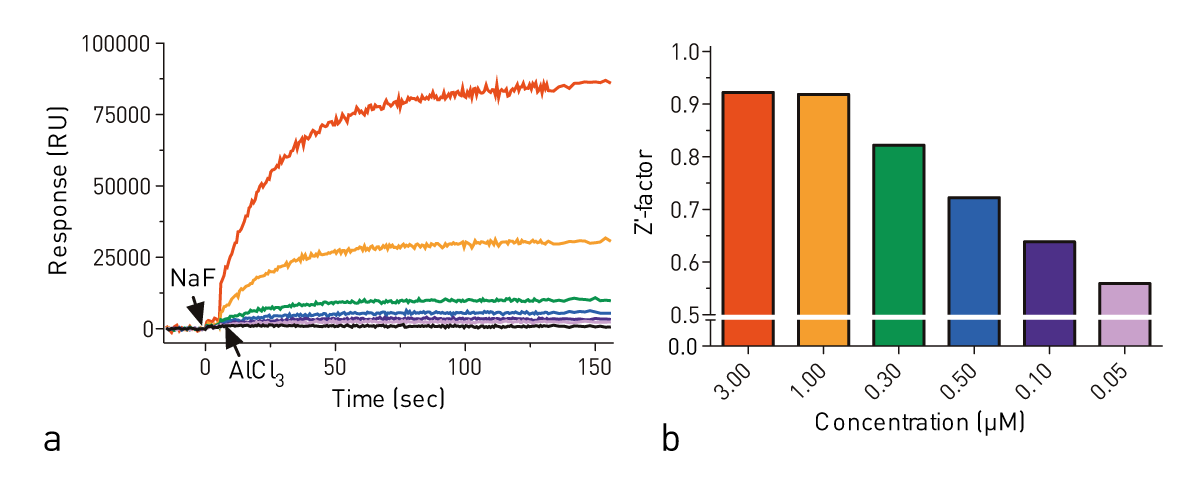
In order to measure the effect of sample concentration on maximal response, we made serial dilutions of Gαi1 from 3 μM to 50 nM in Tris pH 8.0 assay buffer. The most robust response was seen at the highest concentration of Gαi1 tested (Fig. 2, red), but a change in fluorescence was detectable at all concentrations. To compare the quality of the signal for each concentration, a Z’-factor was computed for each concentration.

This calculation accounts for the magnitude of the signal change upon excitation (μplateau - μbaseline) as well as the standard deviation of data collected during the plateau phase (σplateau) and baseline phase (σbaseline). Using the Z’-factor, 3 μM of Gαi1 was seen to have no advantage over 1 μM Gαi1 (i.e., both Z’-factors > 0.9) while the quality of the data decreased at concentrations under 1 μM (not shown).
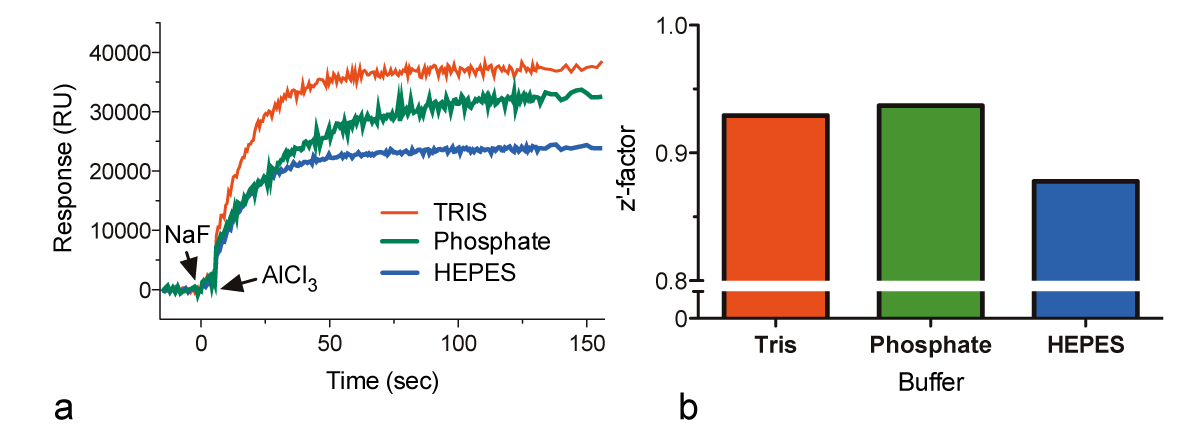
To assess the effect of assay buffer composition on signal intensity, we measured the activation of 1 μM Gαi1 in assay buffer prepared with various common buffer salts (Fig. 3a). The quality of the measurements, as determined by the Z’-factor, was similar for all of the buffers (Fig. 3b) although the maximum signal was observed with Tris assay buffer and the lowest magnitude was observed using HEPES assay buffer.
 To verify that the assay is detecting the rate of Gα activation and is sensitive to changes in this rate, we incubated 500 nM of Gαi1 with 5 μM AGS3Con peptide, a previously described GDI. As expected, the addition of AGS3Con (Fig. 4) dramatically dampened the maximal response of Gαi1, as compared with 500 nM Gαi1 alone.
To verify that the assay is detecting the rate of Gα activation and is sensitive to changes in this rate, we incubated 500 nM of Gαi1 with 5 μM AGS3Con peptide, a previously described GDI. As expected, the addition of AGS3Con (Fig. 4) dramatically dampened the maximal response of Gαi1, as compared with 500 nM Gαi1 alone.
Conclusion
In this application note, we described a robust automated assay system for measuring G-protein a subunit activity. The assay is a sensitive and high-quality means to measure G-protein activation without the use of radiolabeled nucleotides.
Performing the assays with BMG LABTECH´s 96-well plate reader with on-board injectors offers the advantage of automating the assays in triplicate on multiple Gα mutants or multiple modulators of spontaneous GDP release.