PHERAstar FSX
Powerful and most sensitive HTS plate reader
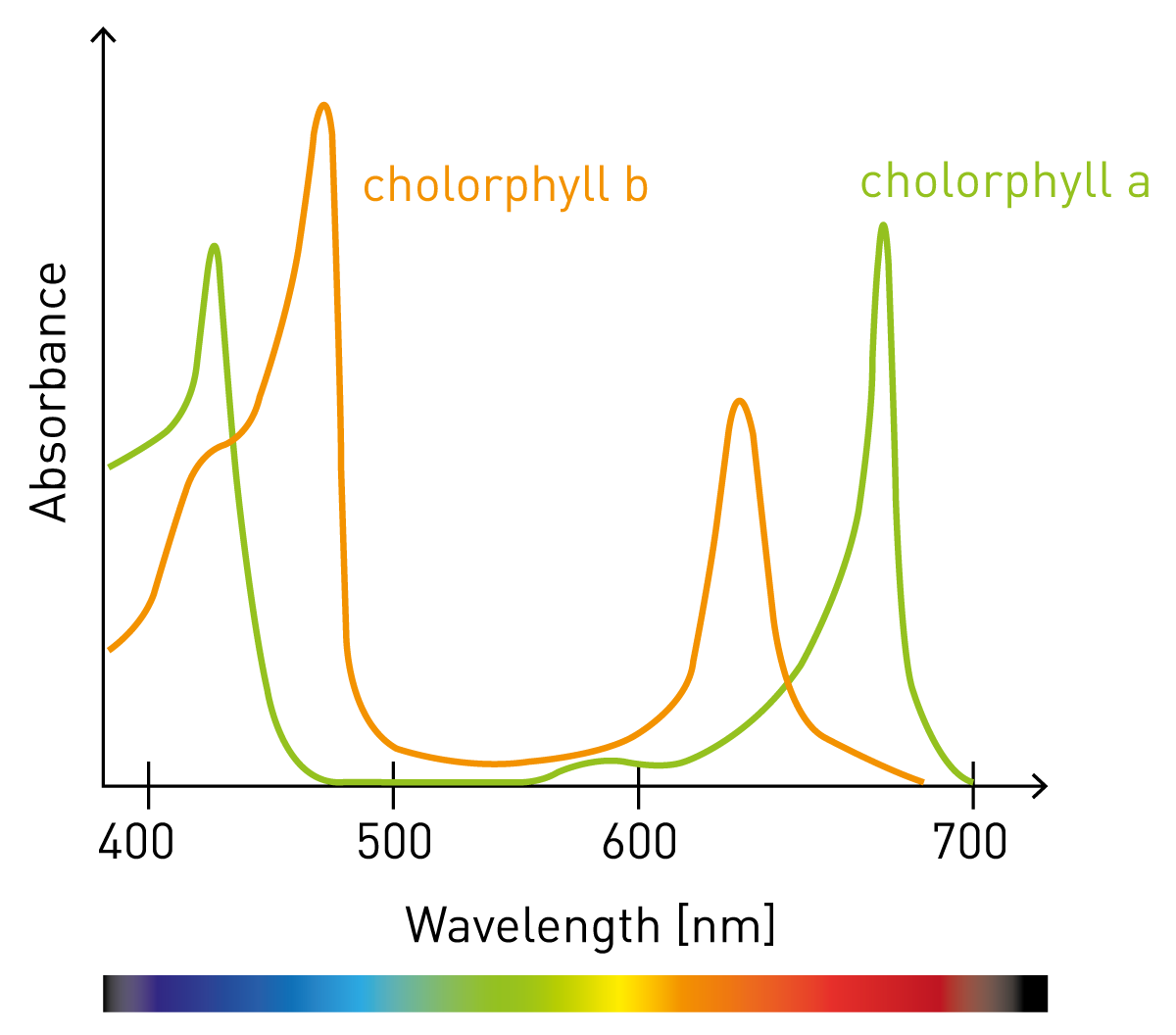
In general, absorbance is a process of light interacting with matter. When light hits something that takes some of the light we talk about absorbance. Of course, the light that is absorbed is not lost; it is transformed to heat or chemical energy as the absorbing molecule gets excited. Absorbance is a physical process we come across every day as we see colors: White light such as sunlight contains all colors of visible light. When it shines on the green grass, the grass absorbs all colors but the green. The green light is reemitted and this is what gives the grass its color.

Detection path in cuvettes and microplates
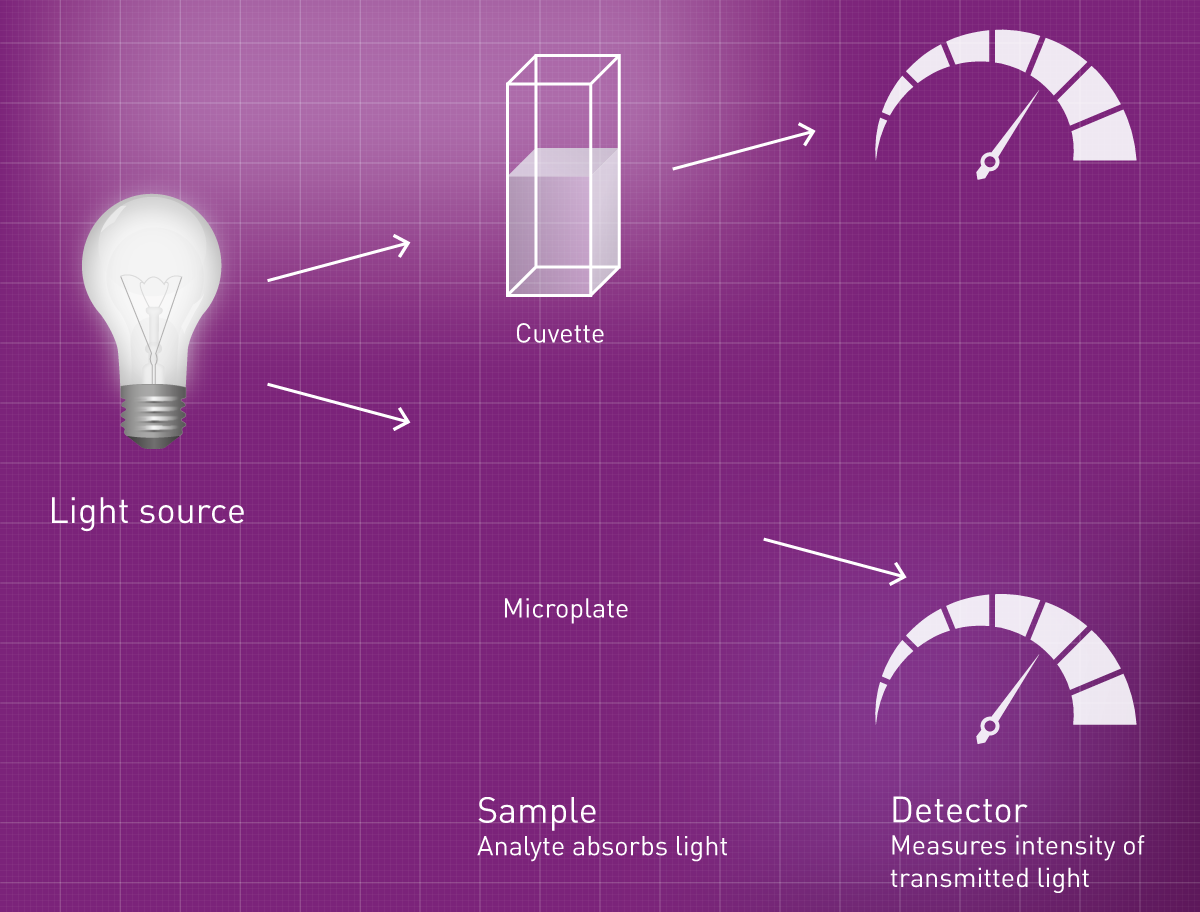
Traditionally, absorbance measurements were performed in a cuvette: A solution with an analyte of known absorbance characteristics is placed into a cuvette. An absorbance reader then determines the absorbance by sending light with known intensity through the sample and detecting the intensity behind the sample. Light that did not make it through to the detector was either absorbed or scattered. The scattered part is determined separately by measuring appropriate blanks and is subtracted from this value to obtain pure absorbance of the substance of interest.
The result of absorbance measurements – transmission, absorbance, and optical density
The portion of the light that is able to pass the sample is also called transmission and is mainly given as percentage (Fig. 2). The more analyte is found in solution, the more light is absorbed by it and the lower is the transmission. The absorbance, however, is the part of the light that was taken up by the analyte. It is the absolute value of the logarithm (to the power of 10) of the transmission1. Here are the mathematical equations and some numbers to explain what is described by transmission and absorbance:
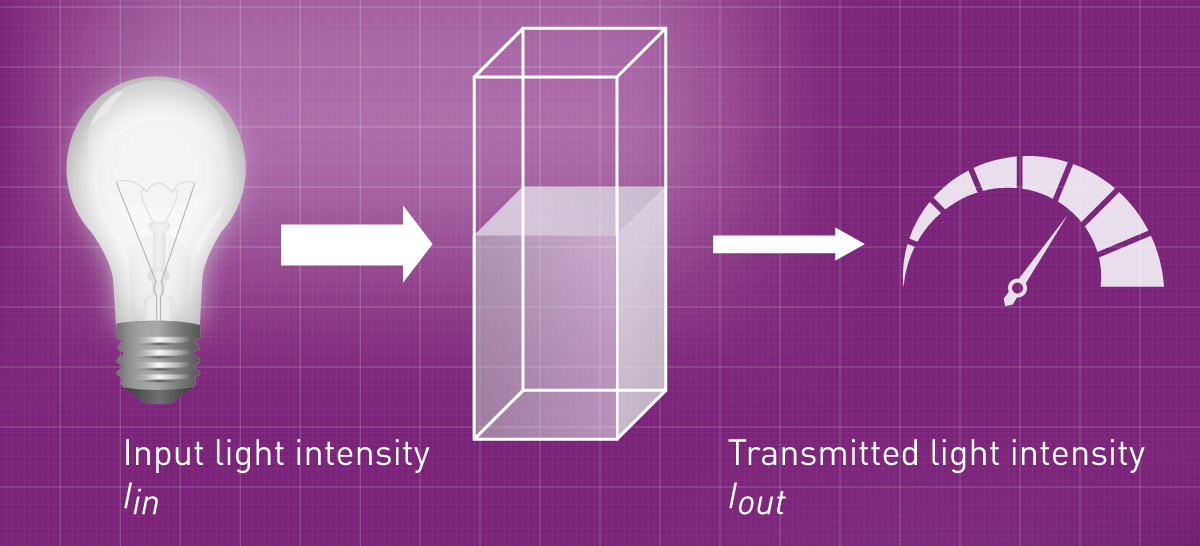
Transmission: T = Iout / Iin
Absorbance: A = -log10T
Table 1: Examples for Transmission and according Absorbance values
| Transmission | Absorbance |
| 0.1 (or 10%) | 1 OD |
| 0.01 (or 1%) | 2 OD |
| 0.001 (or 0.1%) | 3 OD |
Absorbance in chemistry and life sciences - quantify a substance in solution
After performing an absorbance measurement the result is a value given in either transmission or optical density. However, the goal of the measurement is the quantification of a substance in solution, the obvious question is how to convert the signal into the concentration value. Generally, there are two ways: by employing the Beer-Lambert law or by measuring a standard curve in parallel to samples of unknown concentrations.
Beer-Lambert Law
The Beer-Lambert law describes the relation of absorbance, path length and concentration of an absorbing substance:
A=c*d*ε
Changed to c: c=A/(d*ε)
Beer-Lambert law with A – Absorbance, c – concentration, d – path length, ε – extinction coefficient.
It says absorbance is linear to the concentration multiplied by the path length and extinction coefficient2. The path length refers to the length of sample the light has to go through. For instance, in a cuvette the path is standardized to 1 cm. The extinction coefficient is a constant specific for an absorbing substance and a specific wavelength, typically the absorbance maximum of the substance. It provides information on how strongly the specific substance absorbs light at the specific wavelength. As an example, the mass extinction coefficient for bovine serum albumin (BSA) is 0.67 µl*cm-1*µg-1. Accordingly, a solution of 1 µg/µl BSA with a path length of 1 cm has an absorbance of 0.67 OD.
The Beer-Lambert law is very helpful as it allows quantification of absorbing substances without the need to add any other reagents. However, it is limited as outlined below.
Beer-Lambert law can only be used if
Using a standard curve
If any of these criteria do not apply, there is the possibility to indirectly measure analytes and/or using a standard curve. For instance, colorimetric protein quantification assays such as the Bradford assay depend on a substance that increases absorbance in presence of proteins. The increase is measured in a standard curve with known protein concentrations as well as in samples, so that their concentration can be calculated.
Light source
It is self-explanatory: the measurement of light transmission / absorption first requires light. Different light sources can be used for absorbance measurements. They differ in the spectral range they cover, in their intensity and in the stability of the light they emit.
Tungsten Halogen lamps cover the range from 360 nm to more than 1000 nm and are often used due to their cost-effectiveness. Xenon flashlamps, however, cover the spectral region from 220 nm to 1000 nm and cover the UV-range of the spectrum. This makes the detection and quantification of nucleic acids (260 nm) and proteins (280 nm) possible. BMG LABTECH absorbance microplate readers are equipped with a xenon flashlamp for absorbance measurements and hence provide maximum flexibility to measure any wavelength between 220 and 1000 nm, or all the whole spectrum of wavelengths.
Sample
The liquids of interest need to be transferred either to a cuvette or to a microplate in order to measure their absorbance. The material of cuvette and microplate is always clear to ensure maximum light transmission, as absorbance of the solution and not of the material is of interest for the researcher. Still, various materials are common and differ from one to another. Polystyrene is most often used and may be altered to provide specific binding characteristics, either for cell culture or ELISA applications. However, they are non-transmissive in the UV-range which requires the use of special plates of cyclic olefin copolymer to measure absorbance below 400 nm.
Sample volume
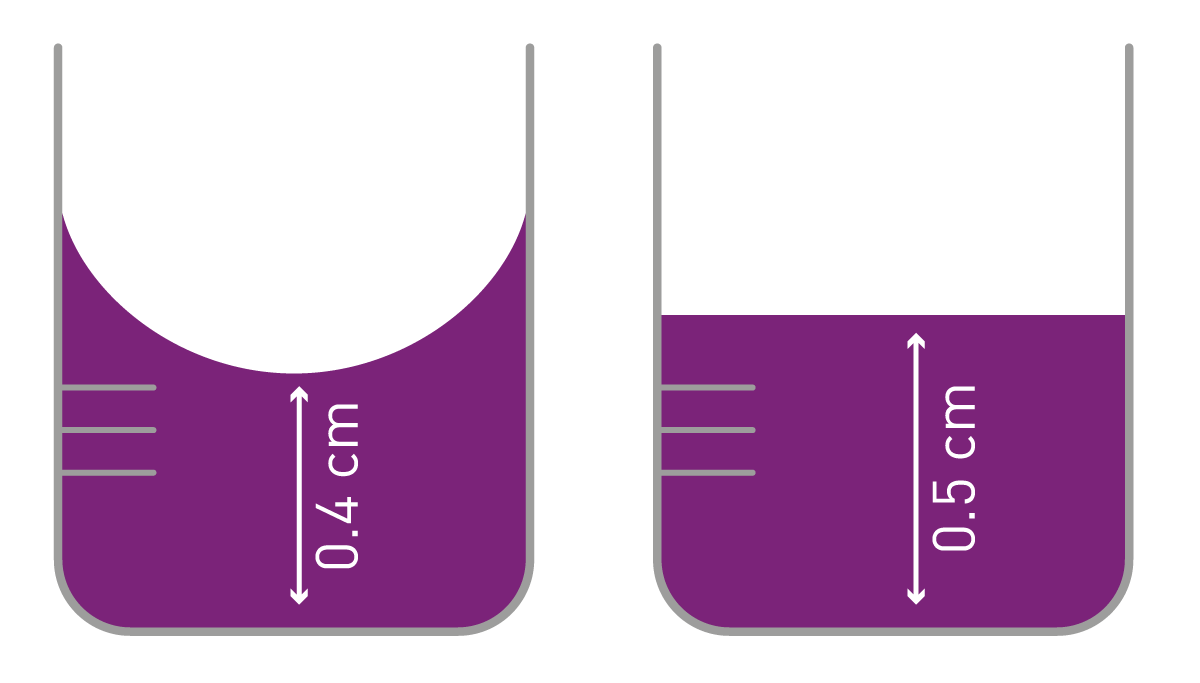
A second aspect to consider is the sample volume that is needed for reliable absorbance measurements. Standard cuvettes require approximately 4.5 ml whereas semi-micro volume cuvettes allow reducing the volume down to 1.5 ml and micro-volume cuvettes get along with 70 µl. The cuvette measurement is horizontal: light is directed from one side into the sample and on the opposite side the transmitted light is detected. Therefore, the geometry of cuvettes provides a standardized path length of 1 cm. This is one of the major differences to microplate measurements as they run vertically (top to bottom or bottom to top). Accordingly, the path length also changes with the sample volume and requires the use of equal volumes for one experiment. Additionally, meniscus effects play an important role for the path length in microplate absorbance measurements. Measurements performed in the middle of a microplate well suffer from a much shorter path in presence of a strong meniscus compared to a measurement with no meniscus. This is of specific importance when applying Beer-Lambert or when having highly variable menisci in an experiment. In aqueous solutions it is possible to correct for meniscus-dependent path length changes by exploiting the absorbance of water (water-peak path length correction).
Standard 96 well plates are typically used with sample volumes ranging between 100 - 300 µl, translating to an approximate path length if 2.9 to 7.4 mm. It might be necessary to further reduce the volume of precious sample without compromising in path length. To this end, half area plates or higher density plates may be used. They provide a smaller well area but the same height as standard 96-well plates and, thus, providing high path lengths with lower volumes.
The appropriate blank
In order to correct absorbance measurements for undesired absorbance from buffer components, from the plate and from light scattering effects a blank is measured in parallel to samples. The appropriate blank contains all components of the assay except from the analyte. Conversely, this means that a PBS or water blank is often insufficient.
Special attention should be paid to particles found in the analyzed samples and blanks. Particles scatter the light and increase the measured absorbance value since less light reaches the detector. As particles move in solution they are detected when they block the light path. When moving out of the detection path the will not be measured. This phenomenon increases the variability of absorbance data. If particles cannot be removed from measured solutions it is recommended to cover a bigger area of the microplate well in the absorbance measurement, a feature all BMG LABTECH devices offer for absorbance measurements.
Detector of absorbance measurements
After the transmitted light passed the sample it needs to be detected. Two different types of detectors are commonly used in microplate readers: either a photomultiplier tube (PMT) or a CCD spectrometer. PMT- based systems require the selection of wavelength before the detection. This is done by optical filters or monochromators that select the desired wavelength before it is guided to the sample. The transmitted light of the desired wavelength then reaches the PMT. The PMT amplifies the signal from the transmitted light and results in a voltage indicative of its intensity. In order to scan absorbance over a range of wavelengths, PMT-based systems use a monochromator that selects the desired wavelength for each point in the spectrum and measures them sequentially.
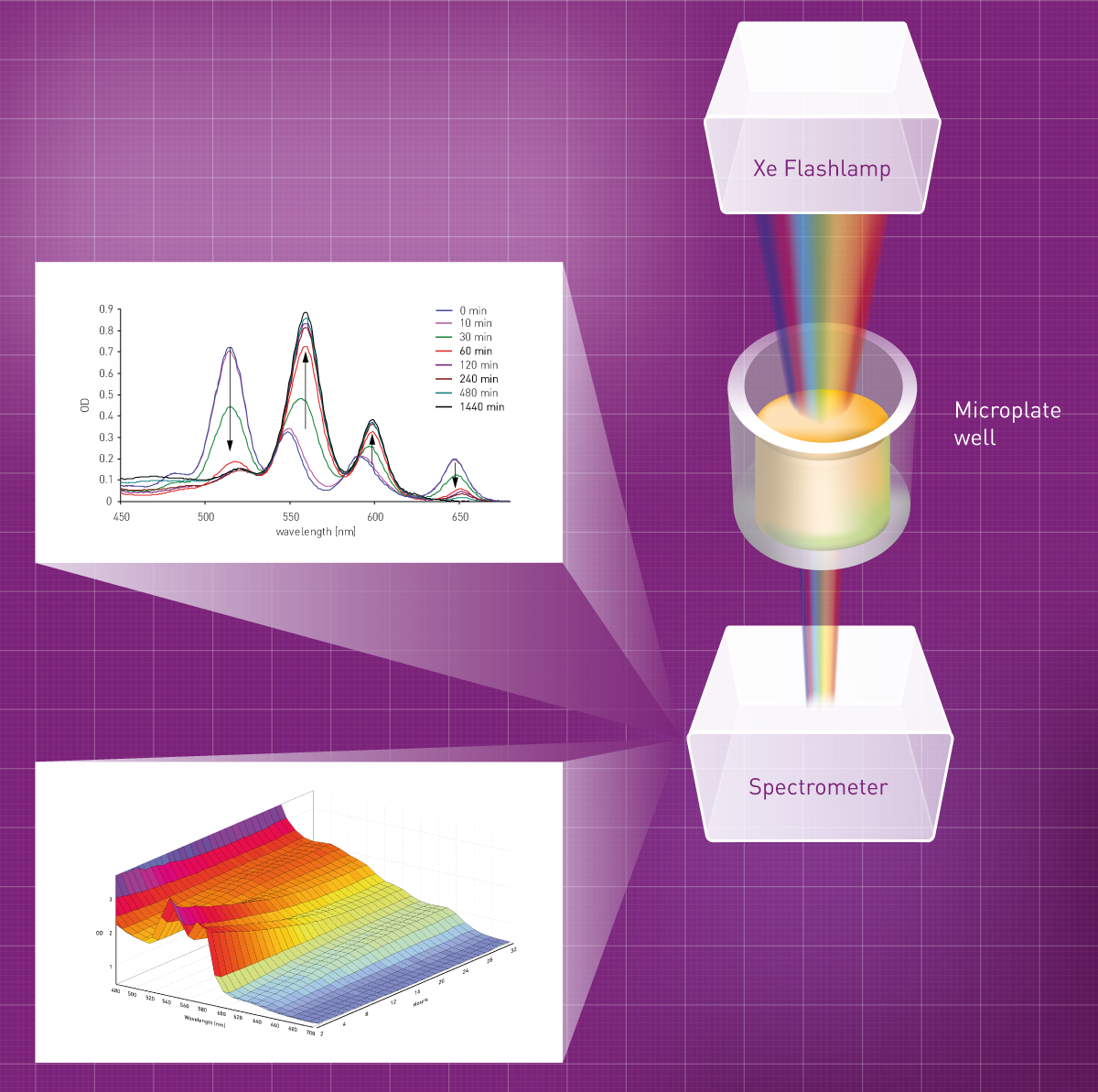
This is the main difference to the second type of absorbance detectors, the spectrometer. It splits the transmitted light into different wavelengths and these are directed onto a CCD detector, capturing the intensity of all wavelengths at once. This way, spectra as well as single or few wavelengths measurements can be acquired in a similar amount of time. For instance, BMG LABTECH instruments employ UV/vis Spectrometer for absorbance measurements that capture a whole spectrum (220 1000 nm) in less than a second.